ISSN : ISSN: 2576-1412
Journal of Applied Microbiology and Biochemistry
Gene Function Expression Profile of Faba bean (Vicia faba) Seeds
Shi Y Yang1*, Michael J Wilkinson2, Yunjie Wang3, Jiang Li3 and Jeffrey G Paull1
1School of Agriculture Food and Wine, The University of Adelaide, Australia
2Pwllpeiran Upland Research Centre Institute of Biological, Environmental and Rural Sciences, Aberystwyth University, UK
3BGI Tech Solutions Co., Ltd, Beishan Industrial Zone, China
- *Corresponding Author:
- Shi Y Yang
School of Agriculture Food and Wine
The University of Adelaide
Waite Campus, Waite Road
Urrbrae, SA 5064, Australia
Tel: +61 (0) 8 8313 7074
E-Mail: shi.yang@adelaide.edu.au
Received Date: June 15, 2017; Accepted Date: June 22, 2017; Published Date: June 29, 2017
Citation: Yang SY, Wilkinson MJ, Wang Y, Jiang Li, Paull JG (2017) Gene Function Expression Profile of Faba bean (Vicia faba) Seeds. J Appl Microbiol Biochem. Vol. 1 No. 3:11
Abstract
Faba bean (Vicia faba L) is one of the important grain crops worldwide and its genome, the largest among grain legumes (approx. 13.4 Gb), has yet to be sequenced. Comprehensive knowledge of genes expressed in the crop's large seeds would not only help drive new genetic improvements in the crop but also aid its future genome characterization. Here, we applied high throughput RNASeq (Quantification) technology to compare gene expression profiles of seeds recovered from six faba bean varieties with divergent agronomic and seed quality attributes. We identified a total of 47,621 Unigenes across all genotypes and a mean count of 38,712 per genotype, total genes length 27605508bp. Comparison between expression levels in lines possessing contrasting phenotypes allowed us to identify candidate genes that may be associated with key traits. In all pairwise comparisons of genotypes, pairwise up-regulated plus down-regulated differences varied between 8,661 and 12,337 genes and co-expressed genes fluctuated between 30,239 and 35,884. Overall, there was a mean of 24.2% genes that were differentially expressed between pairs of genotypes. They were similar of GO profiles generated between the two phenotypic traits (Hydration Capacity and Pea seed-borne mosaic virus (PSbMV) pools and comparison of the GO profiles generated by all pairs of individual genotypes. This is the first comprehensive analysis of gene expression genetic profile on faba bean seeds.
Keywords
RNA-Seq (Quantification); Faba bean (Vicia faba); Seed; Genome assembly; Gene expression profile
Introduction
Faba bean (Vicia faba L) is one of the older crops of the world and its cultivation dates back to small holder Bronze Age farms in the Mediterranean region [1]. Today, it is a global legume grain crop with 4.7 million tonnes produced annually over 3.4 million hectares (FAOSTAT 2015) [2]. However, despite its age and commercial importance, and the fact that it is a diploid species (2n=12), there has been relatively modest progress in developing a good genetic understanding of the crop. In large part this can be attributed to the exceptionally large size of faba bean genome, approximately 13.4 Gb [3], the largest genome in the grain legume family. Assembly of a draft genome of the crop has remained elusive. However, the provision of a detailed transcript profile from the seeds of this species from contrasting phenotypes could provide candidate genes and functional groups implicated in the control of the more important agronomic traits and at the same time, provide a useful platform from which a gene map could be assembled for the species.
RNA-Sequence technology has been used to generate genomewide transcriptome profiles across a wide range of crops including rice [4], maize [5,6] chickpea [7,8] field pea [9,10] and Raphanus sativus [11]. However, the relevant works on faba bean [12,13] have focussed on describing genome-wide expression levels in leaves, stems and T-cells rather than of the large seeds which represents the harvested part of the plant and so holds greatest agronomic interest, and there is no gene expression profiles report for faba bean seeds. Hence, we applied RNA-Seq (Quantification) technology in this study, to discover the gene expression profiles of faba bean seeds from five Australian faba bean varieties and one breeding line with contrasting major seed traits. RNA-Seq (Quantification) is used to analyze gene expression of certain biological objects under specific conditions [14,15]. It is a cost-effective quantification method that produces high reproducibility, high accuracy and wide dynamic range. RNA-Seq can be applied in drug response, biomarker detection, basic medical research, and drug R&D. RNA-Seq can be applied in gene expression analysis, differential gene expression analysis, expression pattern analysis of Differentially Expressed Genes, and Gene ontology classification R&D. The advantages of this technology are as follows: (1) Digital signal and no background noise, can identify sequence differences at the base level; (2) Genes with high or low expression both can be detected; (3) Accurate quantitative results and excellent technology repetition; (4) Both model and non-model organisms can be researched and (5) Analysis of results can be updated in pace with database updating.
Materials and Methods
Five Australian faba bean varieties (Farah, Nura, PBA Rana, PBA Warda, and PBA Zahra) and one breeding line (AF06125) were used for the RNA-Seq (Quantification) technology analysis. These varieties represent the range of diversity among Australian faba bean varieties and include diverse germplasm within their pedigrees [16]. The six genotypes differ for a number of major traits such as Hydration Capacity, time of flowering, seed staining due to Pea seed-borne mosaic virus (PSbMV) and resistance to fungal diseases including Ascochyta blight and rust.
Total RNA extraction
Mature faba bean seeds were ground to powder in liquid nitrogen and dispensed into 500 μL Trizol (Invitrogen, China). After adding 100 μL of chloroform, the mixture was vortexed and chilled on ice for 15 min. The chilled mix was subjected to centrifugation (6500 x g) at 4°C for 30 min. The upper (aqueous) phase was retained, mixed with an equal volume of 70% ethanol (in DEPC H2O). RNA in the solution was then purified using an RNeasy kit (Qiagen, Australia) according to the manufacturer's instructions.
The diagram of the RNA-Seq experimental process shows the steps for the experimental pipeline. Total RNA samples were treated with DNase I (New England Biolabs, China) to degrade any contaminating DNA and then enriched for mRNA with oligo (dT) magnetic beads (Figure 1). The mRNA was fragmented into short fragments (about 200 bp) and cDNA synthesized by random hexamer-primed reverse transcription [17]. Buffer, dNTPs, RNase H and DNA polymerase I were added to synthesize the second strand. The double stranded cDNA was then purified with magnetic beads, end repaired and Ion Proton adaptors ligated (an adapter with barcode and P adapter). Ligated ds cDNAs were size-selected and purified on a 1% (w/v) TAE-agarose gel. Finally, the fragments were enriched by PCR amplification, purified using magnetic beads and dissolved in the appropriate amount of Epstein-Barr solution. During the QC step, Agilent 2100 Bioanaylzer was used to qualify and quantify the sample library. The library products were ready for sequencing on the Ion Proton platform performed by the Beijing Genomics Institute (BGI-Shenzhen).

Figure 1: Diagram of RNA-Seq experimental process.
The Bioinformatics Analysis pipeline shows that the primary sequencing data produced by Ion Proton, called as raw reads, was subjected to quality control (QC) that determined if a resequencing step was needed. After quality control, raw reads were filtered into clean reads which were transformed to fq format, and aligned to the reference sequences at the same time. QC of alignment was performed to determine if resequencing was needed (Figure 2). The alignment data was utilized to calculate distribution of reads on reference genes and mapping ratio. If alignment results passed QC, we proceeded with downstream analysis including gene expression and deep analysis based on gene expression (PCA/correlation/screening differentially expressed genes and so on). Further, we performed deep analysis based on DEGs, including Gene Ontology (GO) enrichment analysis, Pathway enrichment analysis, cluster analysis, protein-protein interaction network analysis and finding transcriptor factors. Here we present the report of the data and result up to Gene Ontology analysis.

Figure 2: Diagram of bioinformatics analysis pipeline.
Faba bean seeds genome assembly
The Trinity method [18] was applied for the de novo assembly of full-length transcripts of these faba bean seeds. Trinity comprises of three sequential software packages (Inchworm, Chrysalis and Butterfly) to process large volumes of RNA-Seq reads in the absence of a reference genome sequence. Each package performs a different function. Inchworm assembles the RNA-Seq data into the unique sequences of transcripts and often produces full-length transcripts for homozygous or dominant isoforms. Inchworm only reports the unique portions of alternatively spliced transcripts. Chrysalis associates the Inchworm contigs into clusters and constructs a de Bruijn graph for each cluster. Each cluster represents the transcriptional diversity for a given gene (or gene family). The package then divides the full read set between these disjoint graphs. Finally, Butterfly traces the path that reads or pairs of reads follow within a graph and so thereby generate full-length transcripts for alternatively-spliced isoforms whilst also isolating any transcripts from paralogous genes.
The file inchworm.K25.L25.DS.fa was the intermediate set while file Trinity.fasta was the final result of Trinity. The parameters of Trinity were as below:
--min_contig_length 100 --min_kmer_cov 3 --inchworm_cpu 8 --bfly_opts '-V 10 --edge-thr=0.05 --stderr' --group_pairs_distance 250 --path_reinforcement_distance 100 --bflyHeapSpaceMax 3G --bflyHeapSpaceInit 1G
Then file Trinity.fasta was taken into further processes of sequence splicing and redundancy removing with sequence clustering software to acquire non-redundant Unigenes as long as possible by TGICL and Phrap softwares to get the final outcome file called All-Unigene.fa. The parameters of TGICL were -l 40 -c 10 -v 25 while the parameters of Phrap were -repeat_stringency 0.95 -minmatch 35 -minscore 35.
The resultant Unigenes were divided into two broad classes: Clusters and Unigenes. Clusters are denoted by the prefix CL followed by the cluster id. Within clusters, there are a number of Unigenes where the similarity between the individual Unigenes is more than 70%. The remainder are deemed singletons, and allocated the prefix Unigene.
Post-assembly analyses
Quality analyses: Sequence saturation analysis was performed to provide an estimate of the coverage of expressed genes detected in the analysis for each genotype. A plotted growth in Unigenes showed that as the absolute number of reads increased the number of detected genes increased. Then, a read distribution analysis across all Unigenes was employed to test for randomness of reads across transcripts. If randomness is good, coverage (Tag counts) across the transcript should be fairly evenly distributed, with neither a strong decline nor accumulation of tags occurring from 5' to 3' end of the transcript.
Gene Expression
Expression levels of individual Unigenes were quantified using the software package Sailfish [19]. This package operates in two sequential phases: indexing and quantification. The index is assembled from a set of reference transcripts and a specified k-mer length, k. The program then estimates maximum likelihood abundance using the Expectation-Maximization (EM) algorithm as a statistical basis to determine which transcripts are isoforms of the same gene. Expression level was measured in reads per kilobase per million mapped reads (RPKM) according to the following formula:
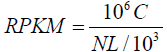
Where C is the number of reads that are uniquely aligned to a specified gene (A), N is the total number of reads uniquely aligned to all genes and L is the length of the specified gene (A) in bases. The RPKM method was selected because it is able to eliminate the influence of different gene length and sequencing level on the calculation of gene expression. This means the calculated gene expression can be directly used for comparing the different expression between samples.
Correlation between samples
Linear correlation of RPKM values was used to assess the robustness of experimental comparisons made between genotypes.
Screening for Differentially Expressed Genes
Screening for differentially expressed genes was based on the Poisson distribution method described by [20] and corrected P-values using the Bonferroni method [21]. Since DEG analysis generates a large multiplicity of problems in which thousands of hypotheses (is gene x differentially expressed between the two groups) are tested simultaneously, correction for false positive (type I errors) and false negative (type II) errors was performed using the False Discovery Rate (FDR) method [22]. We used FDR ≤ 0.001 and the absolute value of Log2Ratio ≥ 1 as the threshold to judge the significance of gene expression difference. Finally, Differentially Expressed Genes (DEGs) were subjected to Gene Ontology (GO) functional analysis.
Results
A similar quantity of clean cDNA reads was obtained from all six genotypes with total reads varying only slightly between the 11.0 M reads for PBA Warda to 13.4 M for AF06125, and with an overall mean across all lines of 12.6 M reads encompassing a mean total read length per genotype of 1,624 Megabase pairs. The GC content of cDNA reads was similarly stable between genotypes, with an overall mean of 45.07% GC across all genotypes varying only slightly between 44.79% (Farah) and 45.84% (PBA Zahra) (Table 1).
Table 1 Data of fababean seeds RNA-Seq (Quantification)It showed the data of faba bean seed RNA- Seq (quantification) obtained from five varieties and one breeding line in this study.
| Sample Name | Clean reads (bp) | Total basePairs | Gene map Rate (%)* | Expressed Gene number | GC content (%) |
|---|---|---|---|---|---|
| AF06125 | 13428227 | 1.7E+09 | 97.59 | 37001 | 44.91 |
| Farah | 12587033 | 1.64E+09 | 97.76 | 40226 | 44.79 |
| Nura | 12892081 | 1.69E+09 | 97.8 | 40434 | 44.67 |
| PBARana | 13287688 | 1.62E+09 | 98.08 | 35814 | 45.09 |
| PBAWarda | 11025828 | 1.48E+09 | 97.61 | 39359 | 45.1 |
| PBAZahra | 12487316 | 1.62E+09 | 98.64 | 39438 | 45.84 |
| Average | 12618029 | 1.62E+09 | 97.91 | 38712 | 45.07 |
Note: *Gene map rate means the total expressed gene number which mapping into the reference genome rate
Faba bean seeds genome assembly
The approach yielded a mean gene classification of 97.9%, with success rates varying within the range 97.6-98.6% between genotypes (Table 1). Unigenes were divided into two broad classes according to the distinctiveness of their RNA sequence, viz: Clusters and Unigenes. In all, we identified 47,621 Unigenes that exceeded the threshold length of 300 bp. There was a near log-linear decay in Unigene size beyond 300bp with a median gene length (N50) of 803 bp, and a maximum length of 6,656. Overall, there were 7,092 Unigenes that exceeded 1 kb in length, 1,350 longer than 2 kb and just 229 above 3 kb (Figure 3 and 4). There was a high level of consistency between genotypes in the number of expressed unigenes inferred by Trinity, with the total number per genotype varying from 37,001 (AF06125) to 40,434 (Nura), and with an overall mean of 38,712 Unigenes per genotype.

Figure 3: Length distribution of all-unigenes of faba bean.

Figure 4: The gene map of faba bean seeds showed total of 47,621 unigenes that exceeded the threshold of 300 bp were identified. Total gene length was 27605508bp with a median gene length (N50) of 803 bp, and a maximum length of 6,656. Overall, there were 7,092 unigenes that exceeded 1 kb in length, 1,350 longer than 2 kb and 229 above 3 kb.
A series of control analyses was undertaken to assess the quality of the RNA-Seq data generated. First, the proportion of unmapped reads was <2.5% for all genotypes surveyed, suggesting that the frequency of aberrant reads must have been extremely low. Second, the coverage of reads across the unigenes identified was evenly distributed, with only the extreme 3' and 5' ends being relatively under-represented (Supplementary Figure S1, Supplementary Table S1). Third, overall correlation between genotypes in their RPKM values and recorded r2 values were above the 0.92 threshold recommended by ENCODE consortium in all pairwise comparisons (Supplementary Table S2). Finally, sequence saturation analyses revealed that Unigene detection was approaching saturation for all genotypes, with estimated total coverage of seed transcriptomes ranging between 86% and 96% (Supplementary Figure S2). Therefore, it is concluded that genome maps were of high quality and provided good coverage of the faba bean seeds map. (RNA sequences data were deposited at National Centre for Biotechnology Information (NCBI) as BioProject ID PRJNA319071 and SRA accession reference number is SRP074308. The faba bean Assembly data were deposited in Figshare: The data DOI is: Digital Object Identifier 10.6084/ m9.figshare.4910039).
The variation in transcript appearance between the six genotypes provided a measure of consistency expected across the crop. In a qualitative sense, there were very few genes that were unique to individual genotypes. Indeed, genotype-specific genes decreased in abundance from a maximum in PBA Zahra (324 genes) to PBA Rana (128 genes), with intermediate genotypes following the order: Farah (292), Nura (219), PBA Warda (176) and AF06125 (173). In all pairwise comparisons of genotypes, the majority of genes were co-expressed between seed transcriptomes (Supplementary Figure S3), with pairwise divergences (upregulated plus down-regulated differences) varying between 8,661 and 12,337 genes, compared with the number of coexpressed genes fluctuating between 30,239 and 35,884. Overall, there was a mean of 24.2% (range 19.2-29.0%) of genes that were differentially expressed between pairs of genotypes. Thus, our results imply that there is relatively modest variation between genotypes in the identity of genes expressed in their seeds but significant differences in the level of expression. Of the genotypes included in the study, the seeds of AF06125 hold particular agronomic interest in that they have outstandingly high hydration capacity (HC) values [23]. The 173 genes specific to this variety were therefore seen as being of interest as potential candidates contributing to this trait.
Differentially expressed genes (DEGs) between varieties
The comparison of gene expression levels between genotypes was aided by the high level of consistency in the total number of RNA reads and genes recovered from each genotype (Table 1). Pairwise comparisons of genotypes for global expression levels revealed relatively large numbers of significantly differentially expressed genes (Table 2). An unrooted tree compiled using the genes that were significantly differentially expressed between genotype pairs revealed little obvious structure except to suggest that PBA Warda possessed a slightly more isolated profile than the other genotypes (Supplementary Figure S4). However, the high degree of consistency in the proportion and identity of differentially expressed genes seen between all pairs of genotypes when compared together (Figure 5; Supplementary Figure S5) suggested global variance among genotypes was both even and relatively modest.
Table 2 Number of Faba bean seeds Differentially Expressed GenesItshowed the number of fababean seed differentially expressed genes (up-regulated and down-regulated) detected in 15 pairs of single variety comparisons and 3 groups of traits comparisons. Group A: lowest hydration capacity genotype; Group B: highest hydration capacity genotype; Group C: intermediate hydration capacity genotype; Group H: low seed staining of Pea seed-borne mosaic virus (PSbMV) genotype and Group I: high seed staining of Pea seed-borne mosaic virus (PSbMV) genotype.
| Pairs/Type | Differentially Expression Gene (Up-regulated) Number | Differentially Expression Gene (Down-regulated) Number |
|---|---|---|
| AF06125-VS-Farah | 11676 | 6819 |
| AF06125-VS-Nura | 11299 | 7053 |
| AF06125-VS-PBARana | 9625 | 8853 |
| AF06125-VS-PBAWarda | 9136 | 7825 |
| Farah-VS-Nura | 8059 | 8794 |
| Farah-VS-PBARana | 7050 | 11220 |
| Farah-VS-PBAWarda | 6947 | 10026 |
| Nura-VS-PBARana | 7285 | 10885 |
| Nura-VS-PBAWarda | 6584 | 9159 |
| PBARana-VS-PBAWarda | 8871 | 8316 |
| PBAZahra-VS-AF06125 | 7906 | 10091 |
| PBAZahra-VS-Farah | 11245 | 8101 |
| PBAZahra-VS-Nura | 10763 | 8497 |
| PBAZahra-VS-PBARana | 9136 | 10314 |
| PBAZahra-VS-PBAWarda | 8744 | 9024 |
| groupA-VS-groupB | 1147 | 319 |
| groupC-VS-groupB | 982 | 174 |
| groupH-VS-groupI | 328 | 175 |

Figure 5: Hierarchical clustering of gene expression for all genotypes. A gene tree is at the left of the figure and each row represents a gene. Expression differences are shown in different colours and red indicates high expression and blue is low expression. Cluster analysis was performed with cluster and java treeview software. The high degree of consistency in the proportion and identity of differentially expressed genes seen in all genotypes suggested global variance among genotypes was both even and relatively modest.
DEGs between phenotypic traits groups
There was a relative evenness of gene presence and DEGs distribution for comparisons of pairs of individual genotypes. While the comparison between the phenotype groups was also similar for the two traits the scale of the variation was much lower than for the individual genotype comparisons. These DEGs were to identify provisional candidates that may be associated with their highly divergent seed features of economic interest.
The genotypes were allocated to three groups according to seed Hydration Capacity (HC), with PBA Zahra showing lowest HC (designated group A), AF06125 being an outlier exhibiting highest HC (designated Group B) and four intermediate genotypes (Farah, Nura, PBA Rana, PBA Warda) comprising Group C. Only a small proportion of genes were differentially expressed between the most contrasting groups, PBA Zahra (Group A) yielding 1,147/47,150 (2.4%) up-regulated and just 319/47,150 (0.7%) down-regulated genes compared to AF06125 (Group B) (Figure 6). Interestingly, comparison of the intermediate genotypes (Group C) with AF06125 produced an extremely similar result with the former exhibiting 982/46,999 (2.1%) up-regulated and 174/46,999 (0.4%) down-regulated genes relative to the latter (Figure 6). Comparison of DEGs generated from the two comparisons revealed a reasonably high level of conservation in the identity of the differentially expressed genes (Supplementary Figure S5), indicating commonality in expression divergence between the high HC genotypes and low/intermediate HC genotypes.

Figure 6: Lowest (Group A) compared to highest (Group B) hydration capacity; intermediate (Group C) compared to highest (Group B) hydration capacity and low (Group H) compared to high (Group I) seed staining due to pea seed-borne mosaic virus. The scatter plots indicated the number of genes that are significantly expressed, up-regulated (orange colour) and down-regulated (blue colour) for one group compared to the other, not significantly expressed in both groups (brown colour).
The genotypes were grouped according to susceptibility to seed staining caused by Pea Seed-Borne Mosaic Virus (PSbMV) infection, with AF06125, PBA Rana and PBA Warda (Group H) all showing low levels of seed staining, compared to the highly staining genotypes of PBA Zahra, Farah and Nura (Group I). This comparison yielded far fewer DEGs, with just 0.7% (328/47,323) genes being up-regulated and 0.4% (175/47,323) being downregulated in the 'susceptible' group (H) (Figure 6).
Gene ontology (GO) genes functional classification
Gene Ontology (GO) functional enrichment analysis integrates cluster analysis of gene expression patterns. GO genes were classified into three clusters based on function, namely Biological Process, Cellular Component and Molecular Function. There was remarkable consistency in the profiles of DEGs identified between the highest HC genotype (Group B) and the other two groups (Figure 7). In the Biological Processes component, metabolic processes represented the most significant GO cluster of DEGs in both comparisons, followed by Cellular Processes and Single-Organism processes. Cell, Cell parts, Organelle and Membrane/Macromolecule complexes dominated DEGs in both comparisons for the Cellular Component. Finally, the Molecular Function component was overwhelmingly dominated in both comparisons by Binding and Catalytic Activity.

Figure 7: Lowest (Group A) compared to highest (Group B) hydration capacity intermediate (Group C) compared to highest (Group B) hydration capacity and low (Group H) compared to high (Group I) seed staining due to pea seed-borne mosaic Virus. WEGO software was used for GO functional classification for DEGs and to understand the distribution of gene functions. GO genes were classified into three clusters: biological process, cellular component and molecular function. In the biological processes component, metabolic processes represented the most significant GO cluster of PEGs, followed by cellular processes and singleorganism processes; cell, cell parts, organelle and membrane/macromolecule complexes dominated for the cellular component; and binding and catalytic activity was dominated in the molecular function component.
The GO profiles were near identical for the comparison of groups according to susceptibility to seed staining caused by PSbMV (Group H vs. Group I), (Figure 7) despite the composition of the two groups being radically different.
Given the close similarity of GO profiles generated between the two phenotypic traits pools, further comparison of the GO profiles was generated for all pairs of different genotypes. There was strong consistency between all pairwise GO profiles that matched those seen in the pooled experiments (Supplementary Figure S6).
Discussion and Conclusion
There are essentially two computational approaches used to reconstruct transcriptomes from cDNA libraries: Map-first and Assembly-first methods [24]. The former initially aligns all reads to a reference genome before merging sequences that overlap the reference genome to form contigs and annotating. The latter starts by assembling overlapping contigs from RNA reads to compile transcripts, potentially allowing the characterisation of a transcriptome in the absence of a reference genome. There have been many studies that have successfully deployed RNASequence approaches to generate global transcriptome profiles [25] often using closely related species to provide a reference genome.
The Trinity method for the de novo assembly of full-length transcripts was applied for faba bean seeds. Trinity contains three sequential software packages (Inchworm, Chrysalis and Butterfly) to process large volumes of RNA-Sequence reads in the absence of a reference genome and has become the most popular approach for the characterisation of RNA-Sequence data of non-model plants [26-32]. However, the approach is not free of problems and it is prudent to perform several quality tests on the RNA-Sequence data and the resultant assembly prior to deeper analysis. The four tests used here all indicated the RNA-Sequence data generated was of high quality for all runs. Specifically, unmapped reads was invariably low <2.5%, read coverage was evenly distributed across the full length of the genome, correlation between RPKM values of individual genotypes were all within the 0.92-0.98 range advocated by the ENCODE Consortium
(https://genome.ucsc.edu/ENCODE/protocols/dataStandards/ ENCODE_RNAseq_Standards_V1.0) and Unigene detection was approaching saturation for all genotypes. These features allowed us to interrogate the data more thoroughly.
In gross terms, the five varieties (Farah, Nura, PBA Rana, PBA Warda, and PBA Zahra) and one breeding line (AF06125) included in this study yielded a total of 47,621 Unigenes across all genotypes and a mean count of 38,712 per genotype. This represents the most comprehensive coverage of faba bean seed mRNA transcripts performed to date. There are three recent publications related to transcriptome analysis of faba bean. The first [10] used pooled samples from leaves, flowers, immature pods, and immature seeds; and seedling-derived roots and shoots from two varieties Icarus and Ascot. RNA sequencing by 454 Roche GS FLX Titanium Technology, generated 41,049 transcripts with annotation, and Unigene number of 18,052. The second by [12] used whole roots, leaves, stems and flowers of one variety Fiord, for RNA sequencing by Illumina platform. This generated 21,297 contigs (17,160 annotated) for V.faba Transfer cell development. The third [13] studied leaves of two faba bean genotypes (29H - resistant to Ascochyta bight and Vf136 - susceptible to Ascochyta blight) following inoculation of Ascochyta fabae. Only 393 and 457 transcripts over- expressed in those two varieties respectively in response to the infection.
Closer examination of our results revealed that the seed transcriptome of faba bean has an unremarkable GC content: higher than Potato (39.9-46.4%), Pea (41.9-44.9% and Chickpea 40.3%, but similar to Arabidopsis (43.4-46.6%), Soybean (45.8- 49.1%), Tobacco (41.8-47.4%) and Tomato (41.7-47.2%), and lower than Rice (54.2-67%), Barley (55.2-66.0%) and Maize (55.8- 67.4%) [33]. Of greater note was the low number of transcripts found to be Unique to a particular genotype. There was a mean frequency of just 0.56% genotype-specific Unigenes among the six genotypes studied (range 0.36% (PBA Rana) to 0.82% (PBA Zahra)). The true value is likely to be even lower than this given that although all libraries were approaching saturation none was completely saturated and so it is possible that rarer transcripts that are universally present may nevertheless occasionally only be detected in one genotype. Thus, there was only modest genotype-unique genetic variation amongst the study set, despite their relatively wide overall genetic variability. This feature could have value in helping to identify candidate genes responsible for traits found only in one of the six genotypes. For example, Hydration Capacity (HC) is a feature that describes the uptake of water during soaking prior to cooking or canning. It is expressed as the percentage of weight gain during a standard time of soaking (usually 16 hours). Water enters the seed via a lacuna in the testa at the raphe, fills the space between testa and the cotyledons and then diffuses into the cotyledons [34]. There are many possible routes by which variation in HC could be mediated. A forward genetics approach to address this problem is made extremely difficult by the large size of the faba bean genome. Similarly, a treatment-control based transcriptomic approach (Hydrated vs. pre-hydrated control comparison) is equally unlikely to yield results because the physical features of the seed causing high HC are almost certainly set during seed maturation and not at the time of processing. In our study, one of the six genotypes (AF01625) possessed markedly raised HC values relative to the other genotypes and yet possessed only 173 genotype-specific Unigenes that could include candidates responsible for this particularly problematic agronomic trait. Clearly, work would be needed to refine the search but the study has at least provided a platform for further work.
In contrast, quantitative comparison of expression revealed relatively large numbers of DEGs between pairs of genotypes and arguably provided a more direct route for enhancing mechanistic understanding of important seed traits. Here, the high degree of consistency in the proportion and identity of differentially expressed genes between all genotype pairs suggested global variance among genotypes was even. The two comparisons made by screening DEGs between the highest HC genotype AF01625 with a group of intermediate genotypes, and with a genotype with lowest HC, which yielded modest numbers of DEGs (1,466 from the former and 1,156 from the latter) although both were far higher than the 173 genotype-specific transcripts identified above and will require more work to identify candidate genes. At first glance, the remarkable consistency in the Gene Ontology profile of these two comparisons could be taken as being mutually supportive. However, as discussed below, caution should be exercised when interpreting GO profiles [35]. The trait of seed staining caused by PSbMV infection, a virus that causes reduced marketable quality of seed in legumes, with faba bean varieties frequently being susceptible to the disease [36,37]. This is a trait where genome size makes it difficult to deploy forward genetics approaches and where concern is not restricted to resistance per se but also includes the absence of symptoms. It therefore provided a useful tool to assess the number of background DEGs expected between equal groups of genotypes and provide a control GO profile of those DEGs identified. In the event, there were smaller numbers of DEGs identified between these pools (1.1%, 503/47,323) than between the two HC pool comparisons (2.5% and 3.1%). A comprehensive set of pairwise comparisons between individual genotypes generated a wide range in the percentage of DEGs detected (19-29% of Unigenes) but also yielded astonishingly stable GO profiles. Consistency between these many GO profiles generated in the present work should be viewed as revealing of the categories of gene attributes that are most and least variable for mature faba bean seeds.
This is the most comprehensive, high quality RNA-Seq data set for mature seeds of the problematic crop (Faba bean), and spanning 6 diverse genotypes. This resource will provide a platform for future efforts to characterise the large genome of the species and to improve mechanistic understanding of the economically important seeds of this species [38]. Comparison of the genotype AF01625 with the other genotypes identified 173 genotype-specific transcripts that could serve as candidates for the important Hydration Capacity trait [39-41].
Acknowledgements
We acknowledge funding from Grains Research & Development Corporation (GRDC) and support from University of Adelaide for the study. Thanks to the faba bean group staff: Ian Roberts, Samuel Catt and Paul Swain for assistance.
We would like to thank Professor Robert Gibson for encouragement on writing the paper. Thanks to Dr Yongle Li, SARDI Gene Function Laboratory for comments on the data analysis.
Author Contributions
S Yang, J Paull, M Wilkinson and Y Wang designed the experiment. J Li carried for all sequences data analysis. Yang wrote the first manuscript, Wilkinson made critical revisions and the manuscript was edited by all authors.
Competing Financial Interests
The authors declare no competing financial interests.
BioProject ID PRJNA319071 and RNA-Seq (Quantification) data are deposited at National Centre for Biotechnology Information (NCBI) gene bank. Reference number is SRA accession: SRP074308. The faba bean Assembly data deposited in Figshare: The data DOI is: Digital Object Identifier 10.6084/m9.figshare.4910039.
References
- Zohary D, Hopf M (1973) Domestication of pulses in the old world: legumes were companions of wheat and barley when agriculture began in the near east. Science 182: 887-894.
- Johnston JS, Bennett MD, Rayburn AL, Galbraith DW, Price HJ, et al. (1999) Reference standards for determination of DNA content of plant nuclei. Am J Bot 86: 609-613.
- Xu H, Gao Y, Wang J (2012) Transcriptomic analysis of rice (Oryzasativa) developing embryos using the RNA-Seq technique. PLoS One 7: e30646.
- Davidson RM, Hansey CN, Gowda M, Childs KL, Lin H, et al. (2011) Utility of rna sequencing for analysis of maize reproductive transcriptomes. The Pla Gen 4: 191-203.
- Kakumanu A, Ambavaram MMR, Klumas C, Krishnan A, Batlang U, et al. (2012) Effects of drought on gene expression in maize reproductive and leaf meristem tissue revealed byrna-seq. PlaPhysiol 160: 846-867.
- Garg R, Patel RK, Tyagi AK, Jain M (2011) De Novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res 18: 53-63.
- Hiremath PJ, Farmer A, Cannon SB, Woodward J, Kudapa H, et al. (2011) Largeâ€ÂÂscale transcriptome analysis in chickpea (Cicerarietinum L.), an orphan legume crop of the semiâ€ÂÂarid tropics of Asia and Africa. Plant Biotechnol J 9: 922-931.
- Sudheesh S, Sawbridge TI, Cogan NO, Kennedy P, Forster JW, et al. (2015) De novo assembly and characterisation of the field pea transcriptome using RNA-Seq. BMC Gen 16: 1-15.
- Kaur S, Pembleton LW, Cogan NO, Savin KW, Leonforte T, et al. (2012) Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Gen 13: 1-12.
- Wu G, Zhang L, Yin Y, Wu J, Yu L, et al. (2015) Sequencing, de novo assembly and comparative analysis of Raphanussativustranscriptome. Front in Pla Sci.
- ArunChinnappa KS, McCurdy DW (2015) De novo assembly of a cotyledon-enriched transcriptome map of Viciafaba (L.) for transfer cell research Front in Pla Sci.
- Ocana S, Seoane P, Bautista R, Palomino C, Claros GM, et al. (2015) Large-Scale Transcriptome Analysis in Faba Bean (Viciafaba L.) under Ascochytafabae Infection. Plos One.
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Meth 5: 621-628.
- Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: A revolutionary tool for transcriptomics. Nat Rev Genet 10: 57-63.
- Kaur S, Cogan N, Forster J,Paull J (2014) Assessment of genetic diversity in faba bean based on single nucleotide polymorphism. Diversity 6: 88-101.
- Wang Y, Chen Q, Chen T, Tang H, Liu L, et al. (2016) Phylogenetic insights into chineserubus (rosaceae) from multiple chloroplast and nuclear dnas. Front in Pla Sci.
- Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, et al. (2013) De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. NatProtoc 8: 1494-1512.
- Patro R, Mount SM, Kingsford C (2014) Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat Biotech 32: 462-464.
- Audic S,Claverie JM (1997) Thesignificance of digital gene expression profiles. Gen Res 7: 986-995.
- Abdi H (2007) Thebonferonni and šidák corrections for multiple comparisons. Encyclopediaof measurement and statistics 3: 103-107.
- Benjamini Y,Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann of Stat, pp:1165-1188.
- Yang SY, James K, Roberts I, Catt S,Paull JG (20-23 Oct 2013) Hydration testing of Australian faba bean (Viciafaba) breeding lines. PBA Inaugural Pulse Conference Adelaide.
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, et al. (2011) Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nature biotechnol 29: 644-652.
- Martin L, Fei Z, Giovannoni J, Rose J (2013) Catalyzing plant science research with RNA-seq. Front Plant Sci.
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29: 644-652.
- Sun X, Zhou S, Meng F, Liu S (2012) De novo assembly and characterization of the garlic (Allium sativum) bud transcriptome by Illumina sequencing. Plant Cell Rep 31: 1823-1828.
- Franssen SU, Shrestha RP, Bräutigam A, Bornberg-Bauer E, Weber AP (2011) Comprehensive transcriptome analysis of the highly complex Pisumsativum genome using next generation sequencing. BMC Gen 12: 1-16.
- Barakat A,DiLoreto DS, Zhang Y, Smith C, Baier K, et al. (2009) Comparison of the transcriptomes of american chestnut (castaneadentata) and chinese chestnut (castaneamollissima) in response to the chestnut blight infection. BMC Plant Biol 9: 1-11.
- Garg R, Patel RK, Jhanwar S, Priya P, Bhattacharjee A, et al. (2011) Gene discovery and tissue-specific transcriptome analysis in chickpea with massively parallel pyrosequencing and web resource development. Plant Physiol 156: 1661-1678.
- Alagna F, D'Agostino N, Torchia L, Servili M, Rao R, et al. (2009) Comparative 454 pyrosequencing of transcripts from two olive genotypes during fruit development. BMC Gen 10: 1-15.
- Lulin H, Xiao Y, Pei S, Wen T,Shangqin H (2012) The first Illumina-based de novo transcriptome sequencing and analysis of safflower flowers. PloS one 7: e38653.
- Carels N, Bernardi G (2000) Twoclasses of genes in plants. Gen 154: 1819-1825.
- Wood JA, Harden S (2006) Amethod to estimate the hydration and swelling properties of chickpeas (Cicerarietinum L.). J of Food Sci 71: E190-E195.
- Huntley RP, Harris MA, Alam-Faruque Y, Blake JA, Carbon S, et al. (2014) A method for increasing expressivity of Gene Ontology annotations using a compositional approach. BMC Bioinformat 15: 155.
- Coutts BA, Prince RT, Jones RAC (2008) Further studies on Pea seed-borne mosaic virus in cool-season crop legumes: responses to infection and seed quality defects. Aus J of Agri Res 59: 1130-1145.
- Sastry KS (2013) Seed-borne plant virus diseases. Springer India.
- Martin LB, Fei Z, Giovannoni JJ, Rose JK (2013) Catalyzing plant science research with RNA-seq. Front Plant Sci 4: 66.
- Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proceedings of the National Academy of Sciences 95: 14863-14868.
- de Hoon MJ, Imoto S, Kobayashi K, Ogasawara N, Miyano S, et al. (2004) Predicting the operon structure of Bacillus subtilis using operon length, intergene distance, and gene expression information.PacSympos on Biocomp 9:276-287.
- Saldanha AJ (2004) Java Treeview—extensible visualization of microarray data. Bioinformat 20: 3246-3248.
- Ye J, Fang L, Zheng H, Zhang Y, Chen J, et al. (2006) WEGO: A web tool for plotting GO annotations. NucAci Res 34: W293-W297.

Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences