Non-specific Subtilisin Producing Isolates KLU119 Cloning and Expression in pET24a Vector
Shwetha S, Advitha P, Rajeswari P and Hariram N*
Department of Biotechnology, Kalasalingam University, Krishnankoil, Tamil Nadu, India
- *Corresponding Author:
- Hariram N
Department of Biotechnology
Kalasalingam University
Krishnankoil 626126, Tamil Nadu, India
Tel: 9443831095
E-mail: n.hariram@klu.ac.in
Received Date: October 10, 2017; Accepted Date: October 22, 2017; Published Date: October 31, 2017
Citation: Shwetha S, Advitha P, Rajeswari P, Hariram N (2017) Non-specific Subtilisin Producing Isolates KLU119 Cloning and Expression in pET24a Vector. J Mol Biol Biotech. Vol.2 No.3:9
Abstract
Subtilisin is a non-specific protease enzyme (Protein digesting enzyme) initially isolated and characterized in an around Kalasalingam University campus from volatile substances. Total 14 isolate was characterized KLU119, KLU-121, KLU- 122, KLU-137, KLU-138, KLU-153, KLU-161, KLU-166, KLU-178, KLU-191, KLU- 211, KLU-221, KLU-243, and KLU-300 based on the zone formation in the solid media for extracellular subtilisin enzyme express. The PCR analysis of all the 14 isolates carried out of them KLU119, KLU161, KLU166 and KLU187 was amplified in specific primer extracellular subtilisin enzyme producing gene. In the present study, KLU119 was further developed a recombinant enzyme, an alkaline serine protease from halo alkaliphilic bacteria, which has been cloned and expressed in DH5α (E. coli) as host. The enzymes BamHI/EcoRI were used for restriction and digestion of the gene 1170 bp. The gene was further subjected to 39 kDa protein expression and analysis. pET24a was used as a potential cloning expression vector. The properties of the enzyme were then compared with the native one. Subtilisin being a protease was studied for its enzyme activity against a tyrosine standard. The maximum concentration of 1.059 Units of enzyme/ml and 18.250 μg protein/ ml of broth was found in KLU119 compare to other isolates. The feasibility of exploiting this recombinant enzyme to various novel applications other than those that already exist was also deduced.
Keywords
Volatile; Primer; Cloning; Digestion; Gene; Recombinant
Introduction
Subtilisin is a non-specific protease (protein digesting enzyme) initially obtained from KLU119 isolate. They belong to subtilases, a group of alkaline serine protease, like all serine proteasesinitiate the nucleophilic attack on the peptide (amide) bond through a serine residue at the active site. They have molecular weights of about 20,000 to 45,000 dalton. They can be obtained from certain types of soil bacteria, for example, Bacillus amyloliquefaciens from which they are secreted in large amounts. Subtilisin are also otherwise known as alcalases, ALKenzyme, bacillopeptidase A, bioprase APL 30, colistinase, subtilisin J, subtilisin S41, subtilisin E, subtilisin BL, genenase I, esperase, savinase 4.0T, savinase, protease S, serine endopeptidase [1].
Extremophiles are often exploited for cloning and over expression of genes in domestic host systems to obtain large quantities of enzymes. The identity of folding and functioning of recombinant protein to the normal protein is necessary and as well as interesting to be investigated. Only a few alkaline proteases are purified and characterized from the halophilic and haloalkaliphilic bacteria so far [2]. Characterization of such haloalkaliphilic enzymes would provide an important clue for the adaptation strategies and stability of the biomolecules under which they can sustain with more than one extremities of NaCl, pH and sometimes temperature [3-5].
In this context, gene cloning from extremophiles into mesophilic bacteria has focused considerable attention. However, as said earlier, only few alkaline serine proteases from halophiles and haloalkaliphiles are purified and characterized due to its instability in the absence of salt. Recombinant DNA technology in conjunction with other molecular techniques is being used to improve, evolve enzymes and for opening new opportunities for the construction of genetically modified microbial strains with the selected biocatalysts. Therefore, cloning of the potential genes coding for different enzymes would be an attractive approval to begin with. Some alkaline protease-encoding bacterial genes have been cloned and expressed in new hosts, the two major organisms for cloning and over-expression being E. coli and B. subtilis [6].
Developments in molecular approaches to improve the cloning and expression of genes leads to enhanced solubilisation of the expressed proteins from halophilic and other extremophilic organisms in heterologous hosts will certainly boost the number of enzyme-driven transformations in chemical, food, pharmaceutical and other industrial applications. This process will add to the prospect of enzyme-driven catalysis [7].
The subtilisin also known as alcalases, which are one of the most important industrial enzymes that are used in many industries, such as detergent, food, pharmaceutical, textile and leather industry. Alkaline proteases that are active in alkaline pH and high temperatures are industrially more important, especially in detergent industry. Although these enzymes has been found in many viruses, fungi and animals. Bacterial alkaline proteases have more importance. Among the bacteria, many species of the genus Bacillus are widely used in industrial production of proteases. Bacillus subtilis subsp. 168as members of this genus that can produce several proteases are also used as a host organism for cloning. Bacillus sp. produces a variety of extracellular proteases at the end of its exponential phase of its growth. Certain other proteases that are expressed in stationary phase of bacterial growth have significant similarity to alcalases, thermitases and pyrolysins [8]. As mentioned above, proteases isolated from bacteria have widespread industrial uses. Here, the protease gene from Bacillus species was cloned for future production of this enzyme for its validation in industrial applications.
In this study, we have developed a recombinant enzyme, an alkaline serine protease from haloalkaliphilic bacteria which has been cloned and expressed in DH5α E. coli as host. The enzymes BamHI/EcoRI were used for restriction and digestion of the gene. The gene was further subjected to protein expression and analysis. pET-24a was used as a potential cloning vector. The properties of the enzyme were then compared with the native one. Subtilisin being a protease was studied for its enzyme activity against a tyrosine standard [9-12]. The feasibility of exploiting this recombinant enzyme to various novel applications other than those that already exist was also deduced. Microorganisms regarded as an important source of proteases because they can be obtained in large quantities using cultural techniques within a shortest possible time by established fermentation methods, and they produce a regular and abundant supply of the desired product. Furthermore, microbial proteins have a longer shelf life and can be stored under less than ideal conditions for weeks without significant loss of activity. Microbial proteases generally have been pointed as to be extracellular in nature and directly express in the fermentation medium [13,14].
Materials and Methods
Bacterial strains and vectors
E. coli strains DH5α and KLU119 were used as bacterial host. The vectors used were plasmid pET-24a.
Sample collection and bacterial isolates
Volatile substances were collected in plastic bags from KLU campus waste volatile substance of water plant. The isolation of bacterial isolate was carried out by serially diluting the volatile samples in sterile water and subsequently plating on the Milk agar base medium and pour plate method. Total number of subtisilin enzyme producing isolates in orderly KLU119, KLU-121, KLU-122, KLU-137, KLU-138, KLU-153, KLU-161, KLU-166, KLU- 18, KLU-191, KLU-211, KLU-221, KLU-243, and KLU-300. All the isolates were incubated in 37°C and one set up of 15% glycerol stock in -70°C.
DNA preparations
Chromosomal DNA was purified as described earlier. Plasmid DNA was prepared by a modified alkaline extraction method. Restriction endonucleases and other enzymes were used according to the supplier’s recommendations. The transformation of E. coli and B. subtilis was made.
Tyrosine assay
Standard Curve Preparation: a) Dilute the Tyrosine Standard to 2.5 mM by adding 25 μL of 100 mM Tyrosine Standard to 975 μL of dH2O. b) Add 0, 2, 6, 12, 18, 24 and 30 μL of Tyrosine Standard into series of wells in a 96-well plate to generate 0, 5, 15, 30, 45, 60, and 75 nmol/well of Tyrosine Standard. c) Adjust the volume to 150 μL/well with dH2O. The diluted Standard can be stored at 4°C for subsequent assays.
Protein analysis by SDS-PAGE
Immune diffusion analysis was according to Ouchterlony. The presence of extracellular proteases was detected on milk agarose plates. SDS-polyacrylamide gels were used to analyze the gene products.
Estimation of extracellular protein
Extracellular protein was estimated by Lowry method. The protein reacts with the Folin Ciocalteau reagent to give a coloured complex. The color formed is due to the reaction of alkaline copper with protein as in the Biuret reaction and the reduction of the phosphomolybdate by tyrosine and tryptophan present in the protein. The intensity of "the colour depends on the amount of these amino acids and thus varies with different proteins.
Amplification of the gene by PCR
The overnight culture of KLU119 in LB broth, the genome of the bacterium was extracted by Qiagen kit. PCR was performed using the forward primer (39mer, 5’ CGC GGATCC ATG AGA AGC AAA AAA TTG TGG ATC AGC TTG 3’) and the reverse primer (39mer, 5’ CGC GAATTC TTA TTG TGC AGC TGC TTG TAC GTT GAT TAA 3’). Two μl of KLU119 extracted genome (50-60 ng) was amplified in a 50 μl reaction mixture containing 2.5 μM of each primers, 5 μl of Taq polymerase, 0.5 mM each deoxyribonucleotide triphosphateand 10X-PCR buffer (containing 2 mM MgCl2). The thermocycling profile was as fallow: initial denaturation at 94°C for 5 min; 35 repeated cycles of 94°C for 1 min, 55°C for 2 min, and 72°C for 3 min; and a final extension of 72°C for 20 min. The PCR product was analyzed using 0.8% agarose gel electrophoresis and digestion with restriction enzymes at 37°C for 1 h.
Construction of the cloning vector containing subtilisin protease gene
The PCR product was extracted from the 0.8% agarose gel using QIA quick Gel Extraction Kit. The amplified gene was inserted into pET24a (+) cloning vector by TA cloning method. For this purpose the insert and vector, DNA ligase (2 μl) and its buffer were added at final volume of 20 μl and incubated at 16°C overnight. Transformation was carried out by heat shocking the E. coli DH5α competent cells prepared by CaCl2 method 39°C for 1 min. After the screening of transformed colonies on LB agar-ampicillin culture medium, plasmid preparation was accomplished by alkaline lysis method. The recombinant plasmids were confirmed with appropriate restriction enzymes.
Results and Discussion
To analyse KLU campus subtilisin enzyme producing organism total fourteen isolates was characterized them from thise isolates KLU119 was brilliantly expression in extracellular enzyme. These isolates were isolated from volatile substance from waste water treatment plant. A number of Bacillus species were grown to late stationary phase and the culture medium was collected to find bacteria that produces subtilisin enzyme. Similarly, one strain was positive, namely a type strain of B. licheniformis, NCIB 6816. Chromosomal DNA from this strain was purified and subsequently used to characterize and clone the gene. To identify the subtilisin gene, we used a synthetic hybridization probe that consisted of 39base pair both forward and reverse primers.
Colony morphology and Gram’s staining
We have to isolate the subtilisin enzyme produced strain in our Kalasalingam University campus. Bacillus sp. KLU119 strain was performed well in subtilisin enzyme production (Figure 1). And basic study was done morphology and grams staining, KLU 119 gram positive rod shaped bacteria (Figures 2a-2c). Further work has been done KLU119 isolate in the agar plate not at all the cross contamination (Figure 2c).

Figure 1: Different subtilisin producing bacterial isolates from KLU.

Figure 2: (a) Morphology of KLU 119 in agar medium (b) Gram’s staining (c) cross contamination of the agar plate.
Strategy of genomic DNA, vector, and PCR product
Isolation of genomic DNA compare to pET24a (+) vector and PCR product in the 0.8% agarose gel (Figure 3).

Figure 3: Primer designed for PCR reaction.
Primer synthesis
Primer synthesis was Forward primer Bam H1 5’ CGC GGATCC ATG AGA AGC AAA AAA TTG TGG ATC AGC TTG 3’ (F-39base pair) and Reverse primer Eco R1 5’ CGC GAATTC TTA TTG TGC AGC TGC TTG TAC GTT GAT TAA 3’ (R-39 base pair) was ligated with BamH1/Eco R1 in pET vector 24a.
An initial experiment was carried out bacterial genomic DNA isolation at cloning the 1.17 kb of BamH1fragment. Genomic DNA was digested with BamH1 and an EcoR1 fragment was separated on preparative 1% agarose gels. DNA fragments in the 1.17 kb range were isolated by the DEAE paper procedure and the fragments were ligated to BamHI/EcoR1 site digested pET24a (+) plasmid. The ligation mixture was used to transform competent E. coli DH5α and the ampicillin resistant colonies were screened for the presence of the subtilisin gene (Figure 3). A similar approach was therefore used to clone the 0.6 kb EcoRI/ HaeIII-fragment. Chromosomal DNA was digested with EcoRI and HaeIII and after separation on agarose gels; fragments between 0.6 and 0.8 kb were purified and ligated to pUC8 digested with EcoRI and SmaI. Several ampicillin resistant clones hybridized to the oligonucleotide mixture, and further analysis of one of the plasmids, pSUB1, showed the presence of a 0.6 kb insert. Partial DNA sequence analysis of the pSUB1 revealed that the insert encoded the C-terminal end of subtilisin, with the orientation of the gene from the EcoRI site towards the HaeIII site (Figure 4).

Figure 4: Schematic diagram of construction and restriction map and sequencing strategy of the gene.
Construction of the cloning vector
After ligation and transformation, the product was spread on LB agar with ampicillin and incubated in 37°C for an overnight to screen the transformed colonies. Seventy one colonies were observed on plate. Plasmid preparation was performed for a number of colonies. To screen the recombinant plasmids, the prepared plasmids were digested by BamH1/EcoRI. Two bands about 1170 bp was observed as shown in Figure 4. The similar results was found in PCR product showed the expected band of 1329 bp. Restriction analysis also confirmed the integrity of the PCR product was carried out by double digestion with NdeI and BamHI (Figure 5).

Figure 5: Restriction and digestions of BamH1/EcoR1 enzyme for PCR product.
Recently no more studies in subtilisin studies not to be done. For the reasons we have been performed in cloning of alkaline protease subtilisin producing orgamim from volatile substance for the first done for 14 isolates. The gene encoding subtilisin Carlsberg from B. licheniformis have been cloned in pBR322 vector that has a 1137 bp open reading frame (ORF) encoding 379 aa. Also, cloning of subtilisin DFE gene from B. amyloliquefaciens DC-4 in pGEM-T plasmid that has a 1146 bp ORF encoding 382 aa. Also B. lentus and B. alkalophilus alkaline protease genes have been cloned and sequenced which both encode 380 aa proteins. Those who are done the work was similar to subtilisin producing wild strains KLU119 was successfully cloned and expressing 39kDa protein in extracellular in liquid medium (Figures 5 and 6). Although another similar results was matched highly antigenic region in the hydrophilic part of the protein including amino acid residues 549 to 795 was successfully cloned and the sequences were confirmed. All nucleotide sequences in the PCR product have 100% homology with the published reference sequence (Figures 6 and 7).

Figure 6: 1% agarose gel (1) gDNA and PCR product (2) cloned 6.3 kb.

Figure 7: Extracellular protein analysis of Clone-119 was expressed in 39 kDa protein in the medium.
Enzyme assay
Not all the isolates were synthesis of non-specific subtilisin enzyme producing microbes in the environment. On the basis of 14 type of isolate was analyzed enzyme assay from waste volatile substance in Kalasalingam University (Table 1). It contains zone of enzyme formation (in cm), PCR amplification, and μg protein/ ml of broth and units/ml/hr of enzyme releases in broth. Among them alkaline protease tyrosine the standard as protease assay was performed to check the activity of the enzyme (Table 1 and Figure 7). The enzyme recommended as 0.07 units/ml solution. However, KLU119 isolates was maximum enzymatic activity in 1.059 units of enzyme released/ml/hr of broth and also protein estimation simultaneously for the concentration 18.250 μg protein/ml of broth (Table 1). The similarly results little less activity expressed in KLU-153, KLU161, and KLU166 isolates was found. The isolate KLU-178 was very less in 0.065 Units of enzyme/ml/hr released and protein of 0.6875 protein/ml of broth. The unit of the enzyme was calculated by the formula and the standard graph is shown in Figure 8.
Table 1 Shown in quantification of enzyme and protein and identification of extracellular enzymereleases in the solid agar medium and find out the PCR amplification of all KLU isolates.
| S.No | Name of the Isolate | Extracellular Subtilisin enzyme in agar medium/PCR |
µg protein/ml of broth | Units /ml of Enzyme | |
|---|---|---|---|---|---|
| Zone in diameter (cm) | PCR amplification | ||||
| 1 | Control (Tyrosine) | - | 0.0 | 0.033 ± 0.002 | |
| 2 | KLU-119 | 2.92 | +++ | 18.250± 0.002 | 1.059 ± 0.002 |
| 3 | KLU-121 | 0.24 | - | 1.500± 0.011 | 0.132 ± 0.012 |
| 4 | KLU-122 | 0.49 | - | 3.065± 0.005 | 0.272 ± 0.001 |
| 5 | KLU-137 | 0.89 | - | 5.5625± 0.006 | 0.491 ± 0.051 |
| 6 | KLU-138 | 0.66 | - | 4.125± 0.011 | 0.367 ± 0.002 |
| 7 | KLU-153 | 2.12 | +++ | 13.25± 0.001 | 0.619 ± 0.005 |
| 8 | KLU-161 | 1.24 | +++ | 7.75± 0.002 | 0.682 ± 0.001 |
| 9 | KLU-166 | 1.12 | +++ | 7.00± 0.001 | 0.617 ± 0.011 |
| 10 | KLU-178 | 0.11 | - | 0.6875± 0.002 | 0.065 ± 0.012 |
| 11 | KLU-191 | 0.21 | - | 1.3125± 0.14 | 0.115 ± 0.020 |
| 12 | KLU-211 | 0.22 | - | 1.375± 0.021 | 0.122 ± 0.021 |
| 13 | KLU-221 | 0.28 | - | 1.75± 0.03 | 0.157 ± 0.110 |
| 14 | KLU-243 | 0.31 | - | 1.9375± 0.11 | 0.170 ± 0.011 |
| 15 | KLU-300 | 1.84 | +++ | 11.500± 0.01 | 0.464 ± 0.111 |
Note: “±” sign indicate 5 replicate

Figure 8: Standard graph of protein estimation.
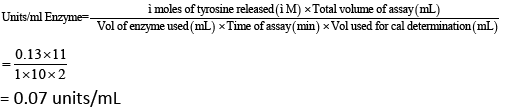
Therefore, the enzyme activity of Recombinant Subtilisin was found to be 0.07 units/mL (Table 1).
Conclusion
In this study we have studied about isolation of different isolate from volatile substances in Kalasalingam University. Total 14 of them were subtilisin producing isolates namely KLU119, KLU- 121, KLU-122, KLU-137, KLU-138, KLU-153, KLU-161, KLU-166, KLU-178, KLU-191, KLU-211, KLU-221, KLU-243, and KLU-300 based on the solid media for synthesising subtilisin enzyme in agar solid media based on the zone formation. Further study has been done PCR amplification of specific primer. However, only five isolates were amplified in this primer, the following isolates were matched KLU119, KLU153, KLU161, KLU166 and KLU300. Interestingly, KLU119 was highly enzymatic activity and also total protein content in this isolates. Further could clone the KLU 119 isolate was synthesis subtilisin enzyme alkaline protease gene in pET24a (+) cloning vector by the fast method of TA cloning kit. This enzyme is a member of subtilisin like protease family with an 1170 bp ORF encoding a390 aa protein was released in extracellular condition (Sequence data not included in this paper).
Acknowledgement
We are sincerely thankful to Director, Chancellor and Vice Chancellor, Kalasalingam University, Krishnankoil, Viruthunagar District, Srivilliputhur-626 126 Tamil Nadu.
References
- Sambrook J, Russel DW (2002) Molecular cloning: A laboratory manual (3rdedn.). Cold Spring Harbor Laboratory Press, New York. pp: 32-33, 116-118.
- Jorgensen PL, Tangney M, Pedersen PE, Hastrup S, Diderichsen B, et al. (2000) Cloning and sequencing of an alkaline protease gene from Bacillus lentusand amplification of the gene on the B. lentus chromosome by an improved technique. Appl Environ Microbiol 66:825-827.
- Jacobs M, Eliasson M, Uhlen M, Flock JI (1985) Cloning, sequencing and expression of subtilisin Carlsberg from Bacillus licheniformis. Nucleic Acids Res 13:8913-8926.
- Peng Y, Yang XJ, Xiao L, Zhang YZ (2004) Cloning and expression of a fibrinolytic enzyme (subtilisin DFE) gene from Bacillus amyloliquefaciens DC-4 in Bacillus subtilis. Res Microbiol 155:167-173.
- Lofdahl S, GussB, Uhlbn M, Lindberg M (1983) Prevalence of β-lactams resistance among Escherichia coli clinical isolates from a hospital in Algiers. Proc Natl Acad Sci 80:697-701.
- Kieser T (1984) Comparative genomics of enterohemorrhagic Escherichia coli O145:H28 demonstrates a common evolutionary lineage. Plasmid 12:19-36.
- Uhlbn M, Guss B, Nilsson B, Gutz F, Lindberg M (1984) Antimicrobial drug resistance: Prediction is difficult, especially about the future. J Bacteriol 159:713-719.
- Hudson L, Hay FC (1976) Practical Immunologysubtilisin-like proteases in plant-pathogen recognition and immune priming: A perspective. J Bacteriol 116:25-32.
- Smith ES, DeLange RJ, Evans WH, Landon M, Markland FS (1968) Plasmodium subtilisin-like protease: Insights into the active-site structure, specificity and function of a pan-malaria drug target. J Biol Chem 243:2184-2191.
- Maniatis T, Fritsch EF, Sambrock J (1982) Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, New York.
- Jacobs M, Eliasson M, Uhlen M, lock JI (1985) Cloning, sequendng and expression of subtilisin Carlsberg from Bacillus lichenifornis. Nucleic Acids Research 13: 245-250.
- Lowry OH, RosebroughNJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265.
- Anson ML (1938) Small RNA mediated repression of subtilisin production in Bacillus licheniformis. J Gen Physiol 22:79-89.
- Acharya MM, Katyare SS (2004) An improved micro method for tyrosine estimation. Med Clin North Am 7: 122-127.

Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences