ISSN : 2471-8548
Journal of Neuropsychiatry
Genetic and clinicopathological contribution of rare ABCA7 mutations in Belgian early onset Alzheimers disease patients
13th World congress on Alzheimers and Dementia
December 06-07, 2018 Amsterdam, Netherlands
Roeck, Sebastiaan Engelborghs, Karin Peeters, Marleen Van den Broeck, Annelies Laureys, Peter P De Deyn, Kristel Sleegers, Patrick Cras and Christine Van Broeckhoven
Center for Molecular Neurology-VIB, University of Antwerp, Belgium Institute Born-Bunge-University of Antwerp, Belgium University of Antwerp, Belgium Hospital Network Antwerp, Belgium Antwerp University Hospital, Belgium
Posters & Accepted Abstracts: J Neurol Neurosci
DOI: 10.21767/2471-8548-C1-003
Abstract
Recent genetic studies suggested an important role for rare deleterious premature termination codon (PTC) mutations in ABCA7 in early-onset Alzheimer’s disease (EOAD). ABCA7 was originally associated with late-onset Alzheimer’s disease (AD) in genome-wide association studies. These ABCA7 mutations are predicted to lead to a loss of functional protein, though their exact mode-of-action is still under investigation. The pathogenicity and segregation patterns of specific mutations and possible modifiers thereof are still poorly understood. We investigated the frequency of ABCA7 PTC mutations in 734 Belgian EOAD patients (mean onset age 61.2±7.0 years) and the clinicopathological features of the mutation carriers. We identified 13 different ABCA7 PTC mutations in 32 carriers (32/734, 4.36 %). Carriers had a mean onset age of 61.7±5.9 (48-70) years. Clinical presentation was predominantly amnestic, except for one patient. No clear distinguishing features were present in the clinical neurological examination or ancillary investigations. A positive first-degree familial history was present in 73.7% (14/19). Neuropathological examination (n=5) showed hallmark AD lesions in 80% (4/5) combined with a pronounced cerebral amyloid angiopathy (CAA). In summary, PTC mutations in ABCA7 are relatively frequent in Belgian EOAD patients, particularly in familial EOAD. Most carriers have a predominant amnestic presentation and their neuropathology shows AD hallmarks in combination with CAA. Continued identification and characterization of ABCA7 mutations is necessary to allow implementation of ABCA7 screening in clinical practice and genetic counselling. Also, ABCA7 PTC carriers might represent an important genetic subtype of AD and more knowledge might improve genetic diagnosis, risk prediction and development of targeted therapeutics.
Biography
E-mail:
liene.bossaerts@uantwerpen.vib.be
Google Scholar citation report
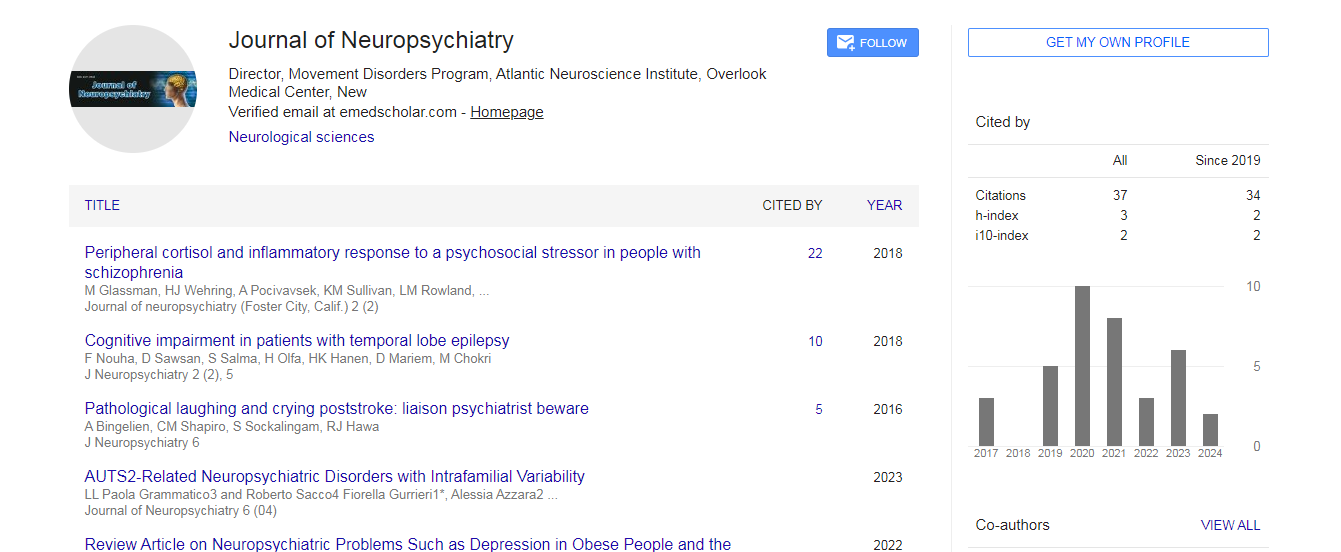
Citations : 37
Journal of Neuropsychiatry received 37 citations as per Google Scholar report
Abstracted/Indexed in
- Google Scholar
- China National Knowledge Infrastructure (CNKI)
- Secret Search Engine Labs
- Euro Pub
Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences