Dynamic Study Oxidation of Methane to Methanol on ds Zone of Transition Metal VIIIB (M@Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt) Catalysts
Hongxia Liu*, Zizhong Liu, Yanhong Zhang and Yuminqiqige Xinxue
Chemistry and Environment Science College, Inner Mongolia Key Laboratory of Green Catalysis, Inner Mongolia Normal University, P.R China
- *Corresponding Author:
- Hongxia Liu
Chemistry and Environment Science College
Inner Mongolia Key Laboratory of Green
Catalysis, Inner Mongolia Normal University, P.R China.
Tel: 0471-4392519
Fax: +86-471-4392124
E-mail: liuhx@imnu.edu.cn
Received date: April 04, 2018; Accepted date: April 17, 2018; Published date: April 21, 2018
Citation: Liu H, Liu Z, Zhang Y, Xinxue Y (2018) Dynamic Study Oxidation of Methane to Methanol on ds Zone of Transition Metal VIIIB (M@Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt) Catalysts. J Mol Cell Biochem. Vol.2 No.1:3.
Abstract
The related energy and multi-channel oxidation of methane to methanol reaction potential energy surface under the ds zone of transition metal catalysts and its dynamic characterization have been investigated with density functional calculations. The geometries were fully optimized with the B3PW91 level. The calculated results shown that the main pathway oxidation of methane to methanol reaction under the ds zone of transition metal VIIIB (M@Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt) catalysts can give the main product P1 (CH3OH+O). We calculated the rate constant of the main reaction pathway, the calculated dynamic characterization indicating that the rate constants increase significantly as the temperature increase. According to the dynamic results and the energetically intermediates and transition states involved in the dominant paths, the reaction is expected to be occurred the most rapidly under the catalysis of VIIIB transition metal. According to our calculation, the title reaction is exothermic reaction under the catalyst of VIIIB transition metal, and it is a thermodynamically feasible reaction. The theoretical reference data on searching new catalysts to catalytic oxidation of methane will be offered. These theoretical results are of important value in theory and application for find new catalysts. The research is of great theoretical and experimental value.
Keywords
Oxidation; Potential energy; Methane; Transition metal catalysts; Temperature; Transition states; Exothermic reaction
Introduction
Methane is a very stable and high symmetry molecular. It can form strong regular tetrahedron symmetry structure by four equivalent C-H through the sp3 hybridization of carbon atoms. Its thermodynamic performance is stable, and the C-H bond energy is as high as 439 kJ/mol-1. It is difficult to activate, and usually need to reaction under high temperature and high pressure. Compared to the reactants, the product is easily activated and make the product selectivity difficult to control.;
Methanol as the basic chemical raw material, can easily turn into an important chemical raw materials and fuels such as olefins, aromatics, and taking methane as the main component of natural gas reserves. This reaction, if it can achieve large-scale. Industrial production, will greatly help mankind to get rid of its dependence on oil. In the catalytic process, however, due to its usually involves an expensive antioxidant, corrosive or reaction medium [1-8], these media are not suitable for large-scale commercial and is still an unsolved problem. Although in the gas phase under mild conditions, methane can be directly into methanol by molecular oxidation, but the process is either stoichiometric (and therefore need a water extraction steps) [9-15], or is too slow and low yield rate and unrealistic [16]. However, it has not been reported that an effective catalyst can be used to effectively use natural gas to convert methane directly into methanol. Therefore, it is important to develop catalysts that can selectively oxidize methane. Recently, the study of Shan et al. proposed two ways to catalyze the oxidation of methane (CH4) into methanol and acetic acid: 1.CH4 produces methanol further oxidation as acetic acid under H2 and CO.2.CH4 is directly oxidized into methanol under the action of O2 and CO2 [17]. Yet large-scale industrial applications remain a difficult practical problem. Apparently, the correlative reactions about methane are very important.
Doped metals have been shown to be effective in improving the physical and chemical properties of oxides [18,19]. After doping, the charge state of the oxide body ion is changed, the charge balance is disturbed, and the excess charge is generated. Therefore doping affect subject oxide electrical, optical properties and chemical reaction mechanism, weakening the crystal chemical bonds within the [20], enhance the adsorption on the surface of the [21-22], is helpful to activate the reconciliation from molecular [23]. Doping of metals tends to enhance the activity of the reaction in the material. In this study, we hope to achieve effective catalytic oxidation of methane by doping transition metal.
Considering the potential importance and the rather limited information, we carry out a detailed theoretical study on the potential energy surface (PES) of the methane oxidation to methanol reaction to (1) provide the elaborated addition channels on the oxidation of methane to methanol reaction PESs; (2) give a deep insight into the mechanism of the methane oxidation to methanol reactions; (3) calculate the rate constant of the methane oxidation to methanol. We hope our work will provide some valuable fundamental insights into oxidation of methane.
Material and Methods
Computational details
All calculations are carried out using the GAUSSIAN03 program packages [24]. The geometries of all the reactants, products, intermediates, and transition states are optimized using the hybrid density functional B3PW91/Lanl2dz method [25,26]. As is known, full electron calculations for transition metal atoms consume too much time, so it is necessary to introduce the relativistic effective core potential (RECP) to describe the inner core electrons. The 5s25p65d106s1 outermost valence electrons of the transition metal atom are described through the Lanl2dz base set [27,28]. The stationary nature of structures is confirmed by harmonic vibrational frequency calculations; i.e., equilibrium species possess all real frequencies, whereas transition states possess one and only one imaginary frequency. To confirm that the transition states connect designated intermediates or products, intrinsic reaction coordinate (IRC) calculation is carried out at the B3PW91 level. The rate constant of the ratecontrolling step along the main reaction channel was calculated among the temperature range 100–1000 K, which considers the small curvature tunnel effect cor-rection (SCT) [29,30].
Results
The optimized structures of the important stationary points of the oxidation of methane to methanol reaction are shown in Figures 1-10. For our convenient discussion, the energy of reactants R is set to be zero for reference. By means of the transition states and their connected isomers or products, a schematic potential energy surface (PES) of the most relevant reaction pathways for the CH4+O2+M(M@Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt) reaction is plotted in Figures 1-10.

Figure 1:A schematic potential energy surface of the CH4+O2→CH3OH+O reaction obtained at the B3PW91 + ZPE level.

Figure 2: A schematic potential energy surface of the CH4+O2+Fe→CH3OH+O+Fe reaction obtained at the B3PW91 + ZPE level.

Figure 3: A schematic potential energy surface of the CH4+O2+Co→CH3OH+O+Co reaction obtained at the B3PW91 + ZPE level.

Figure 4: A schematic potential energy surface of the CH4+O2+Ni→CH3OH+O+Ni reaction obtained at the B3PW91 + ZPE level.

Figure 5: A schematic potential energy surface of the CH4+O2+Ru→CH3OH+O+Ru reaction obtained at the B3PW91 + ZPE level.

Figure 6: A schematic potential energy surface of the CH4+O2+Rh→CH3OH+O+Rh reaction obtained at the B3PW91 + ZPE level.

Figure 7: A schematic potential energy surface of the CH4+O2+Pd→CH3OH+O+Pd reaction obtained at the B3PW91 + ZPE level.

Figure 8: A schematic potential energy surface of the CH4+O2+Os→CH3OH+O+Os reaction obtained at the B3PW91 + ZPE level.

Figure 9: A schematic potential energy surface of the CH4+O2+Ir→CH3OH+O+Ir reaction obtained at the B3PW91 + ZPE level.

Figure 10: A schematic potential energy surface of the CH4+O2+Pt→CH3OH+O+Pt reaction obtained at the B3PW91 + ZPE level.
The related energy
Data on formation enthalpies constitute an excellent means to establish whether theoretically predicted phases are likely to be stable, and such data may serve as a guide to evaluate possible reaction routes. For the exploration of the thermodynamic feasibility of accessing these compounds from the elements (eqn (1)) we have also computed the total energies for CH4+O2+M (M@ Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt) in their ground state structures with full geometry optimization. The adsorption enthalpies for CH4+O2+M (M@Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt) were calculated from the difference in the total energy which are summarized in Table 1. The results establish unambiguously that (eqn (1)) expresses endothermic process for CH4+O2+M (M@Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt) adsorption.
CH4+O2+M→R(CH4+O2+M) (1)
| Enthalpies of formation (DH/kcal mol-1) | |||
|---|---|---|---|
| A | CH4+O2 | - | - |
| DH/kcal mol-1 | 0 | - | - |
| A | CH4+O2+Fe | CH4+O2+Co | CH4+O2+Ni |
| DH/kcal mol-1 | -100.8 | -77.4 | -83.3 |
| A | CH4+O2+Ru | CH4+O2+Rh | CH4+O2+Pd |
| DH/kcal mol-1 | -105.6 | -74.4 | -46.8 |
| A | CH4+O2+Os | CH4+O2+Ir | CH4+O2+Pt |
| DH/kcal mol-1 | -118.8 | -81.2 | -71.5 |
Table 1: Calculated enthalpies of formation (DH/kcal mol-1) according to eqn (1) for the adsorption CH4+O2+M(M=Fe,Co,Ni,Ru,Rh,Pd,Os,Ir,Pt) compounds.
The formation energy for the prototypical CH4+O2+M is labled in Table 1 which indicating that CH4+O2+M is a thermodynamically stable phase at ambient conditions. This has already been established by a series of experimental and theoretical studies. Our estimated large positive values for the enthalpy of formation for the CH4+O2+M series also suggest that it might be possible to oxidation of methane to methanol as stable phases.
Reaction potential energy surface
On the PES for the reaction CH4+O2, the hydrogen-abstraction can form intermediate IM via the transition state TS1. Subsequently, the intermediate IM can occure OH-abstraction to give product P (CH3OH+O) with the barrier TS2. In view of the two barriers, it is impossible to overcome the barrier height for transition state TS1 and TS2 which the energy barrier is too high.
As shown in Figure 1, H-abstraction from the CH4 via TS1 leading to intermediate IM. For the transition state TS1 in Figure 1, the breaking C-H bond is elongated by 43% compared to the C-H regular bond length in CH4, while the forming H–O bond is 7% than the H–O regular bond length. The elongation of the breaking bond is larger than that of the forming bond, indicating that TS1 is product-like, so this reaction will proceed via ‘‘late” transition states as expected for the endothermic reactions. This late character is in keeping with Hammond’s postulate. A schematic potential energy surface of the CH4+O2 reaction obtained at the B3LYP/6-311+G(d,p) + ZPE level is plotted in Figure 1. The ZPE-corrected energy of TS1 (37.2 kcal/mol) is higher than the reactants, which is due to the fact in the present study. As can be seen from the PES, the H-abstraction reaction pathway is endothermic, with the value of 0.8 kcal/mol at the B3LYP/6- 311+G(d,p) level. Secondly, the 1,2-OH-abstraction can occur to form product P (CH3OH+O), which may via transition state TS2. This abstraction-reaction need to overcome 42.0 kcal/mol. As shown in Figure 1, the O-O bond will be broken as 1.346 Å. The oxidation of methane to methanol reaction can be described as:
Path 1: R → IM → P (CH3OH+O)
Compared to the CH4+O2 reaction, the CH4+O2+M (M@Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt) reaction mechanism is the same. The energy barrier of the transition state is all decreased except Co, and the energies of all products are negative which means they are all endothermic reactions. It is shown that the energy variation of the transition state is consistent with the DE variation. It can conclude that the TS1a-j are product-like, this H-shift reaction will proceed via ‘‘late” transition states as expected for the endothermic reactions. When the title reaction catalyzed by M (M@Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt), we can see that the energy barrier of the transition state decreased except Co in Figures 1-10.
In the previous sections, we have obtained oxidation of methane to methanol reaction channels (Path 1) for the CH4+O2+M (M@ Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt) reaction. In the fifth and sixth cycle, with the the number of extra nuclear electrons increase, the energy barrier of the reaction became bigger and bigger as the formation enthalpies increase. The fourth cycle of Fe, Co, Ni do not confirm to this rule.
Kinetics
In this section, we performed the dynamic calculation for the reaction CH4+O2+M (M@Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt). The calculation of the thermodynamic functions ΔH and ΔS at B3PW91 level was used for obtaining the rate constant results of the dominant channel in the 100-1000 K temperature range. Results are listed in Tables 2-11.
| T(K) | Δr≠sm(J K mol-1) | Δr≠Hm(kJ/mol) | k(s-1) |
|---|---|---|---|
| 100 | -83.584 | 153.601 | 5.2 × 10-73 |
| 200 | -91.872 | 152.459 | 1.0 × 10-32 |
| 298 | -93.006 | 152.209 | 1.8 × 10-19 |
| 400 | -91.918 | 152.595 | 1.6 × 10-12 |
| 500 | -90.299 | 153.325 | 1.9 × 10-8 |
| 600 | -88.713 | 154.200 | 1.1 × 10-5 |
| 700 | -87.299 | 155.114 | 1.1 × 10-3 |
| 800 | -86.082 | 156.025 | 3.4 × 10-2 |
| 900 | -85.040 | 156.908 | 5.3 × 10-1 |
| 1000 | -84.149 | 157.762 | 4.8 |
Table 2: The thermodynamic functions ΔH and ΔS at B3PW91 level and the rate constants of the CH4+O2→IM channel in the 100-1000 K temperature range.
| T(K)
|
Δr≠sm(J K mol-1) | Δr≠Hm(kJ/mol) | k(s-1)
|
|---|---|---|---|
| 100 | -0.021 | 39.507 | 4.8 × 10-9 |
| 200 | -1.657 | 39.276 | 1.8 × 102 |
| 298 | -2.105 | 39.168 | 6.5 × 105 |
| 400 | -2.117 | 39.165 | 4.9 × 107 |
| 500 | -2.109 | 39.171 | 6.5 × 108 |
| 600 | -2.197 | 39.118 | 3.8 × 109 |
| 700 | -2.397 | 38.992 | 1.3 × 1010 |
| 800 | -2.682 | 38.777 | 3.5 × 1010 |
| 900 | -3.025 | 38.485 | 7.6 × 1010 |
| 1000 | -3.402 | 38.125 | 1.4 × 1011 |
Table 3: The thermodynamic functions ΔH and ΔS at B3PW91 level and the rate constants of the CH4+O2+Fe→IM channel in the 100-1000 K temperature range.
| T(K) | Δr≠sm(J K mol-1) | Δr≠Hm(kJ/mol) | k(s-1) |
|---|---|---|---|
| 100 | 7.355 | 78.575 | 4.6 × 10-29 |
| 200 | 6.883 | 78.530 | 2.9 × 10-8 |
| 298 | 8.891 | 79.040 | 2.5 × 10-1 |
| 400 | 11.560 | 79.972 | 1.2 × 103 |
| 500 | 13.983 | 81.062 | 1.9 × 105 |
| 600 | 16.087 | 82.218 | 6.0 × 106 |
| 700 | 17.887 | 83.387 | 7.5 × 107 |
| 800 | 19.435 | 84.548 | 5.2 × 108 |
| 900 | 20.782 | 85.688 | 2.4 × 109 |
| 1000 | 21.958 | 86.807 | 8.5 × 109 |
Table 4: The thermodynamic functions ΔH and ΔS at B3PW91 level and the rate constants of the CH4+O2+Co→IM channel in the 100-1000 K temperature range.
| T(K) | Δr≠sm(J K mol-1) | Δr≠Hm(kJ/mol) | k(s-1) |
|---|---|---|---|
| 100 | 1.686 | 69.812 | 8.7 × 10-25 |
| 200 | -0.707 | 69.468 | 2.7 × 10-6 |
| 298 | -1.331 | 69.324 | 3.7 × 100 |
| 400 | -1.322 | 69.327 | 6.3 × 103 |
| 500 | -1.272 | 69.350 | 5.1 × 105 |
| 600 | -1.322 | 69.321 | 9.8 × 106 |
| 700 | -1.481 | 69.214 | 8.3 × 107 |
| 800 | -1.728 | 69.032 | 4.2 × 108 |
| 900 | -2.029 | 68.775 | 1.5 × 109 |
| 1000 | -2.372 | 68.449 | 4.2 × 109 |
Table 5: The thermodynamic functions ΔH and ΔS at B3PW91 level and the rate constants of the CH4+O2+Ni→IM channel in the 100-1000 K temperature range.
| T(K) | Δr≠sm(J K mol-1) | Δr≠Hm(kJ/mol) | k(s-1) |
|---|---|---|---|
| 100 | -4.004 | 11.783 | 9.0 × 105 |
| 200 | -9.590 | 10.958 | 1.8 × 109 |
| 298 | -12.740 | 10.192 | 2.2 × 1010 |
| 400 | -14.393 | 9.624 | 8.2 × 1010 |
| 500 | -15.359 | 9.190 | 1.8 × 1011 |
| 600 | -16.079 | 8.796 | 3.1 × 1011 |
| 700 | -16.694 | 8.395 | 4.6 × 1011 |
| 800 | -17.263 | 7.972 | 6.3 × 1011 |
| 900 | -17.799 | 7.515 | 8.1 × 1011 |
| 1000 | -18.313 | 7.024 | 9.9 × 1011 |
Table 6: The thermodynamic functions ΔH and ΔS at B3PW91 level and the rate constants of the CH4+O2+Ru→IM channel in the 100-1000 K temperature range.
| T(K) | Δr≠sm(J K mol-1) | Δr≠Hm(kJ/mol) | k(s-1) |
|---|---|---|---|
| 100 | -7.858 | 59.064 | 1.1 × 10-19 |
| 200 | -13.77 | 58.211 | 5.0 × 10-4 |
| 298 | -16.397 | 57.575 | 7.0 × 101 |
| 400 | -17.715 | 57.121 | 3.4 × 104 |
| 500 | -18.51 | 56.766 | 1.3 × 106 |
| 600 | -19.133 | 56.422 | 1.5 × 107 |
| 700 | -19.686 | 56.065 | 8.9 × 107 |
| 800 | -20.217 | 55.668 | 3.4 × 108 |
| 900 | -20.723 | 55.232 | 9.7 × 108 |
| 1000 | -21.225 | 54.759 | 2.2 × 109 |
Table 7: The thermodynamic functions ΔH and ΔS at B3PW91 level and the rate constants of the CH4+O2+Rh→IM channel in the 100-1000 K temperature range.
| T(K) | Δr≠sm(J K mol-1) | Δr≠Hm(kJ/mol) | k(s-1) |
|---|---|---|---|
| 100 | -8.004 | 126.613 | 5.8 × 10-55 |
| 200 | -12.439 | 125.974 | 1.2 × 10-21 |
| 298 | -14.288 | 125.528 | 1.1 × 10-10 |
| 400 | -15.138 | 125.236 | 6.0 × 10-5 |
| 500 | -15.648 | 125.011 | 1.4 × 10-1 |
| 600 | -16.079 | 124.769 | 2.5 × 101 |
| 700 | -16.498 | 124.498 | 1.0 × 103 |
| 800 | -16.916 | 96.357 | 1.1 × 106 |
| 900 | -17.338 | 123.826 | 1.5 × 105 |
| 1000 | -17.761 | 123.421 | 8.8 × 105 |
Table 8: The thermodynamic functions ΔH and ΔS at B3PW91 level and the rate constants of the CH4+O2+Pd→IM channel in the 100-1000 K temperature range.
| T(K) | Δr≠sm(J K mol-1) | Δr≠Hm(kJ/mol) | k(s-1) |
|---|---|---|---|
| 100 | -2.782 | -4.657 | 4.0 × 1014 |
| 200 | -3.067 | -4.707 | 4.9 × 1013 |
| 298 | -3.816 | -4.891 | 2.8 × 1013 |
| 400 | -4.561 | -5.151 | 2.3 × 1013 |
| 500 | -5.251 | -5.463 | 2.1 × 1013 |
| 600 | -5.941 | -5.842 | 2.0 × 1013 |
| 700 | -6.632 | -6.291 | 1.9 × 1013 |
| 800 | -7.318 | -6.806 | 1.9 × 1013 |
| 900 | -7.983 | -7.373 | 1.9 × 1013 |
| 1000 | -8.632 | -7.985 | 1.9 × 1013 |
Table 9: The thermodynamic functions ΔH and ΔS at B3PW91 level and the rate constants of the CH4+O2+Os→IM channel in the 100-1000 K temperature range.
| T(K) | Δr≠sm(J K mol-1) | Δr≠Hm(kJ/mol) | k(s-1) |
|---|---|---|---|
| 100 | 5.544 | -7.998 | 6.1 × 1016 |
| 200 | 3.632 | -8.295 | 9.5 × 1014 |
| 298 | 1.841 | -8.736 | 2.6 × 1014 |
| 400 | 0.556 | -9.18 | 1.4 × 1014 |
| 500 | -0.372 | -9.595 | 1.0 × 1014 |
| 600 | -1.125 | -10.01 | 8.1 × 1013 |
| 700 | -1.787 | -10.435 | 7.1 × 1013 |
| 800 | -2.381 | -10.885 | 6.4 × 1013 |
| 900 | -2.941 | -11.357 | 6.0 × 1013 |
| 1000 | -3.469 | -11.859 | 5.7 × 1013 |
Table 10: The thermodynamic functions ΔH and ΔS at B3PW91 level and the rate constants of the CH4+O2+Ir→IM channel in the 100-1000 K temperature range.
| T(K) | Δr≠sm(J K mol-1) | Δr≠Hm(kJ/mol) | k(s-1) |
|---|---|---|---|
| 100 | 3.845 | 35.149 | 1.4 × 10-6 |
| 200 | -0.904 | 34.411 | 3.8 × 103 |
| 298 | -6.121 | 33.119 | 4.7 × 106 |
| 400 | -10.544 | 31.58 | 1.7 × 108 |
| 500 | -14.071 | 30.001 | 1.4 × 109 |
| 600 | -17.041 | 28.37 | 5.5 × 109 |
| 700 | -19.606 | 26.702 | 1.4 × 1010 |
| 800 | -21.874 | 25.005 | 2.8 × 1010 |
| 900 | -23.903 | 23.28 | 4.7 × 1010 |
| 1000 | -25.74 | 21.541 | 7.1 × 1010 |
Table 11: The thermodynamic functions ΔH and ΔS at B3PW91 level and the rate constants of the CH4+O2+Pt→IM channel in the 100-1000 K temperature range.
Rate constants were calculated as follows:
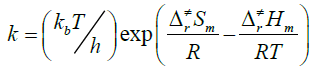 (2)
(2)
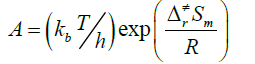 (3)
(3)
Where the type of ΔrHm and ΔrSm are respectively for the standard molar activation enthalpy and entropy of reaction system. A is the frequency factor, and kb is Boltzmann constant.
It is seen from Table 2, it shows that the rate constant k of this reaction increases significantly as the temperature increase.
Among the range of 100–1000 K, we find that the theoretical rate constants are slightly positive temperature dependence at 100– 1000 K. So it is favorable to the most probable reaction at higher temperature [31-33]. On the other hand, compared among these kinds of reactions, the rate constant of CH4+O2+M is the biggest one which is suitable with the analysis of the PESs. According to our calculation, the singlet atom can catalyze the reaction better.
Discussion
Therefore, we hope our present calculation may represent a useful model to understand the mechanism and provide valuable information for the title reaction, and to deeply understand the mechanism of the title reaction. Meanwhile, it will provide theoretical guidance for us to find more effective catalysts.
Conclusion
We have presented a detailed investigation on the related energy and CH4+O2 reaction potential energy surface under the catalysis of M (M@Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt) and its dynamic characterization using a detailed quantum chemical method. The following important conclusions are obtained:
(1) The calculation shows that the large negative formation enthalpy of CH4+O2+M (M@Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt) is consistent with the fact that it has already been catalyzed by experiments. The prediction of CH4+O2 reaction under the catalysis of VIII B (Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt) with large negative formation enthalpy hopefully will inspire and guide catalysis efforts in this direction.
(2) The multi-channel oxidation of methane to methanol reaction potential energy surface under the catalysis of M (M@Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt) is performed to explore the reaction mechanism. The general potential energy surface information is obtained at the B3PW91 level. Our calculation shows that the oxidation of methane to methanol channel is obtained. The H-shift and OH-abstraction reaction can give the main product P (CH3OH+O+M (M@Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt)). And CH4+O2+M (M@Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt) reaction occurs mainly in the high-temperature range which plays a more important role as the temperature increase. The present theoretical studies may provide useful information on the reaction mechanism
(3) From the thermodynamic study, in the fifth and sixth cycle, with the number of extra nuclear electrons increase, the energy barrier of the reaction became bigger and bigger as the formation enthalpies increase. The fourth cycle of Fe, Co, Ni do not confirm to this rule.
(4) The dynamic characterization show that the theoretical rate constants are slightly positive temperature dependence at 100–1000 K. The rate constant k of this reaction increases significantly as the temperature increase. The analyses consistently support the notion that the VIII B transition metal can catalyze the reaction better. Until now, there are no experimental studies available to verify our results, but we hope to motivate experimentalists to measure the rate constant.
Acknowledgements
The project was supported by the Higher educational Scientific research projects of Inner Mongolia (NJZY030), the Natural Science Foundation of Inner Mongolia (2012MS0218), the Talent Development Foundation of Inner Mongolia, the Inner Mongolia Normal University Research Fund Project (KYZR1119), the Natural Science Foundation of Inner Mongolia (2014BS0206), and the Scientific research project of the Inner Mongolia colleges and Universities (NJZY030).
References
- Hammond C, Forde MM, Rahim A, Hasbi M, Thetford A, et al. (2012) Direct catalytic conversion of methane to methanol in an aqueous medium by using copperâ€ÂÂpromoted Feâ€ÂÂZSMâ€ÂÂ5. Angew Chem Int Ed 51: 5129-33.
- Hammond C, Dimitratos N, Jenkins RL, Lopez-Sanchez JA, Kondrat SA, et al. (2013) Elucidation and evolution of the active component within Cu/Fe/ZSM-5 for catalytic methane oxidation: from synthesis to catalysis. ACS catalysis 3: 689-699.
- Liu CC, Mou CY, Yu SS, Chan SI (2016) Heterogeneous formulation of the tricopper complex for efficient catalytic conversion of methane into methanol at ambient temperature and pressure. Ener Environ Sci 9: 1361-1374.
- McFarland E (2012) Unconventional chemistry for unconventional natural gas. Science 338: 340-342.
- Goldshleger NF, Tyabin MB, Shilov AE, Shteinman AA (1969) Activation of saturated hydrocarbons-deuterium-hydrogen exchange in solutions of transition metal complexes. Russian J Phys Chem 43: 1222.
- Periana RA, Taube DJ, Gamble S, Taube H, Satoh T, et al. (1998) Platinum catalysts for the high-yield oxidation of methane to a methanol derivative. Science 280: 560-564.
- Chepaikin EG, Bezruchenko AP, Leshcheva AA, Boyko GN, Kuzmenkov IV, et al. (2001) Functionalisation of methane under dioxygen and carbon monoxide catalyzed by rhodium complexes: oxidation and oxidative carbonylation. J Mol Catal A Chem 169: 89-98.
- Periana RA, Mironov O, Taube D, Bhalla G, Jones CJ (2003) Catalytic, oxidative condensation of CH4 to CH3COOH in one step via CH activation. Science 301: 814-818.
- Groothaert MH, Smeets PJ, Sels BF, Jacobs PA, (2005) Schoonheydt RA. Selective oxidation of methane by the bis (μ-oxo) dicopper core stabilized on ZSM-5 and mordenite zeolites. J Am Chem Soc 127: 1394-1395.
- Sushkevich VL, Palagin D, Ranocchiari M, Van Bokhoven JA (2017) Selective anaerobic oxidation of methane enables direct synthesis of methanol. Science 356: 523-527.
- Grundner S, Markovits MA, Li G, Tromp M, Pidko EA, et al. (2015) Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol. Nat Commun 6: 7546.
- Narsimhan K, Michaelis VK, Mathies G, Gunther WR, Griffin RG, et al. (2015) Methane to acetic acid over Cu-exchanged zeolites: mechanistic insights from a site-specific carbonylation reaction. J Am Chem Soc 137: 1825-1832.
- Starokon EV, Parfenov MV, Pirutko LV, Abornev SI, Panov GI (2011) Room-temperature oxidation of methane by α-oxygen and extraction of products from the FeZSM-5 surface. J Phys Chem C 115: 2155-2161.
- Beznis NV, Van Laak AN, Weckhuysen BM, Bitter JH (2011) Oxidation of methane to methanol and formaldehyde over Co–ZSM-5 molecular sieves: Tuning the reactivity and selectivity by alkaline and acid treatments of the zeolite ZSM-5 agglomerates. Microporous Mesoporous Mater 138: 176-183.
- Shan J, Huang W, Nguyen L, Yu Y, Zhang S, et al. (2014) Conversion of methane to methanol with a bent mono (μ-oxo) dinickel anchored on the internal surfaces of micropores. Langmuir 30: 8558-8569.
- Narsimhan K, Iyoki K, Dinh K, RomaÃŒÂÂn-Leshkov Y (2016) Catalytic oxidation of methane into methanol over copper-exchanged zeolites with oxygen at low temperature. ACS central science 2: 424-429.
- Shan J, Li M, Allard LF, Lee S, Flytzani-Stephanopoulos M (2017) Mild oxidation of methane to methanol or acetic acid on supported isolated rhodium catalysts. Nature 551: 605.
- McFarland EW, Metiu H (2013) Catalysis by doped oxides. Chem Rev 113: 4391-427.
- Nilius N (2015) Exploring routes to tailor the physical and chemical properties of oxides via doping: an STM study. J Phys Condens Matter 27: 303001.
- Kim HY, Lee HM, Pala RG, Shapovalov V, Metiu H (2008) CO oxidation by rutile TiO2 (110) doped with V, W, Cr, Mo, and Mn. J Phys Chem C 112: 12398-12408.
- Mammen N, Narasimhan S, De Gironcoli S (2011) Tuning the morphology of gold clusters by substrate doping. J Am Chem Soc 133: 2801-2803.
- Shao X, Prada S, Giordano L, Pacchioni G, Nilius N (2011) Tailoring the shape of metal Adâ€ÂÂparticles by doping the oxide support. Angew Chem Int Ed 50: 11525-11527.
- Cui Y, Shao X, Baldofski M, Sauer J, Nilius N, et al. (2013) Adsorption, activation, and dissociation of oxygen on doped oxides. Angew Chem Int Ed 52: 11385-11387.
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, et al. (2003) gaussian 03, Gaussian. Inc., Pittsburgh, PA. 2003.
- Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A Gen Phys 38: 3098-3100.
- Perdew JP, Wang Y (1992) Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev B Condens Matter 45: 13244-13249.
- Hay PJ, Wadt WR (1985) Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J Chem Phys 82: 270-283.
- Hay PJ, Wadt WR (1985) Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J Chem Phys 82: 299-310.
- Lu DH, Truong TN, Melissas VS, Lynch GC, Liu YP, et al. (1992) POLYRATE 4: A new version of a computer program for the calculation of chemical reaction rates for polyatomics. Comput phys commun 71: 235-262.
- Liu YP, Lynch GC, Truong TN, Lu DH, Truhlar DG, et al. (1993) Molecular modeling of the kinetic isotope effect for the [1, 5]-sigmatropic rearrangement of cis-1, 3-pentadiene. J Am Chem Soc 115: 2408-2415.
- Liu HX, Wang Y, Yang L, Liu JY, Gao H, et al. (2009) CH3NHNH2 + OH reaction: mechanism and dynamics studies. J Comput Chem 30: 2194-2204.
- Luo J, Jia X, Gao Y, Song G, Yu Y, et al. (2011) Theoretical study on the kinetics of OH radical reactions with CH3OOH and CH3CH2OOH. J Comput Chem 32: 987-997.
- Zhang H, Zhang GL, Liu JY, Sun M, Liu B, et al. (2010) Theoretical studies on the reactions CH3SCH3 with OH, CF3, and CH3 Radicals. J Comput Chem 31: 2794-2803.

Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences