Cancer Incidence in Relation to Species Size: The Peto Paradox in the Light of the Theory of Epigenetic Carcinogenesis
Patrick A Riley*
Totteridge Institute for Advanced Studies, London, United Kingdom
- *Corresponding Author:
- Patrick A Riley
Emeritus Professor of Cell Pathology
University College London, United Kingdom
Tel: (+44) 20 8445 5687
E-mail: p.riley@ucl.ac.uk
Received Date: August 28, 2018 Accepted Date: October 9, 2018 Published Date: October 16, 2018
Citation: Riley PA (2018) Cancer Incidence in Relation to Species Size: The Peto Paradox in the Light of the Theory of Epigenetic Carcinogenesis. J Med Oncol. Vol.1 No.3:11
Abstract
A two-stage model of carcinogenesis, based on the generation of a defect in the vertical transmission of epigenetic information, is outlined. The essential property of the model exhibits an age-related increase in cancer incidence proportional to the number and proliferation rate of the population of stem cells of different tissues. In the light of this, the paradoxical similarity of cancer incidence rates in animals of differing size (such as man and elephants) is discussed in relation to the possible significance of a proofreading mechanism in diminishing the transmission frequency of epigenetic error.
Keywords
Epigenetics; DNA methylation; Carcinogenesis; p53; Elephants
Introduction
A longstanding difficulty in explaining the mechanism of carcinogenesis in terms of the accumulation of somatic mutations has been the need to invoke a mutator phenotype and the fact that the necessary mutations occurred according to the proliferation rate of the tissue. These difficulties are overcome by postulating that the significant error leading to cancer is due to deranged epigenetic inheritance. The development of multicellular organisms entails the distinct specification of cell types with specialised functions. Despite possessing an identical genomic sequence differentiated cells exhibit substantially different profiles of gene expression, and preservation of the developed pattern requires these cellular identities to be conserved during later cell divisions. The role of epigenetic mechanisms is to provide this stable and heritable pattern.
The methylation of cytosine residues within CpG dinucleotides has profound effects on gene expression and it was proposed by Holliday & Pugh [1] and Riggs [2] that this constituted the basic mechanism for generating the different gene expression profiles essential for normal development. The faithful somatic inheritance of the established DNA methylation patterns is due to a “maintenance” mechanism which depends on a methylation enzyme (DNMT 1) that preferentially recognizes hemi-methylated DNA. The details of the whole process whereby the epigenetic pattern is established and maintained involve many other processes including the incorporation of histone variants, and posttranslational modifications of histones which affect chromatin structure. However, the fundamental process is orchestrated by the inherited pattern of DNA methylation as demonstrated by the elimination of differentiated cells in DMNT 1-knockout animals [3].
In eukaryotes, the basic unit of chromatin is the nucleosome which consists of 1.65 turns of DNA wrapped around an octamer of histones that include two copies of the core histones H2A, H2B, H3, and H4 [4]. During mitosis the structure of the nucleosomes is broken down and the DNA released and after the DNA has been replicated the nucleosomal structure is reassembled. The details of the processes involved in the replisome are not fully understood but some of the pre-existing histones are reutilised in addition to the synthesis of new histones. The reassembly of the nucleosomes is guided by the pattern of DNA methylation [5]. Initial copying of the DNA methylation pattern is carried out by DNMT 1 on hemimethylated DNA [6] but there is evidence that the methylation process is completed after the reassembly with the histone components has begun [7]. The entire process of reconstitution of the chromatin structure is complex and subsumes many interactive events [8].
Epigenetic Carcinogenesis
Recently there has been considerable interest in the role of epigenetic mechanisms in cancer [9-12] and it has been proposed that carcinogenesis is the outcome of error-prone transmission of the epigenetic information in proliferating stem cells [13-15]. In terms of the standard two-stage model of carcinogenesis, the origin of this error-prone epigenetic transmission is posited to result from a somatic mutationinduced defect affecting DNA methyltransferases, histone modifying enzymes and factors implicated in reassembly of nucleosomes. Moreover, as in all replicating systems, it is highly probable that an important component of the epigenetic transmission mechanism is a proofreading process which checks the fidelity of the transmitted pattern of DNA methylation and eliminates cells with defective epigenetic copying. It has been suggested that the p53 associated apoptosis mechanism performs such a function [16-18] and it has been proposed that this is an important carcinogenic target since p53 has been shown to be frequently inactivated in cancer cells [19,20].
Failure of fidelity in the copying of the DNA methylation pattern would lead to disturbances of the pattern of gene expression and derangement of the chromatin architecture with resultant widespread genetic instability (CIN) [21]. Hence, cells manifesting this abnormality will give rise to clones with a diversifying range of structural and functional abnormalities. This pathogenesis is consistent with the range of diagnostic features present in cancer cells-such as abnormal mitoses, deranged chromosome pattern, bizarre structure etc.
Since the occurrence of epigenetic error is restricted to mitosis this pathogenic process predicts that the probability of the incidence of cancer will be a function of stem cell proliferation. Thus the tissue-specific variation in cancer incidence would be expected to be related to the number of stem cells and their mean rate of proliferation [22]. Tissues in which mitosis is absent (such as CNS), or rare (such as striated muscle), will not develop cancer, whereas tissues with high proliferation rates (such as epithelia) will have a raised incidence as observed [23]. By a similar argument the cancer risk will be modified by factors that affect the stem cell proliferation rate such as inflammation, hormones and age [24,25].
It is possible to summarize the events envisaged in the foregoing discussion as follows:
1. At each division the cells at risk (tissue stem cells) may be subject to random errors which result in faulty epigenetic copying.
2. In general, faulty epigenetic copying will be detected and the affected cells eliminated by a gatekeeper mechanism.
3. However, if the gatekeeper mechanism is inactivated, each time a stem cell with a defective proofreading mechanism divides, the faulty epigenetic copying goes undetected and the resulting modified pattern of gene expression includes properties that may endow the cell and its progeny with malignant characteristics.
Two-stage Carcinogenesis
This basic idea is consistent with the accepted pathological division of carcinogenesis into two phases respectively known as initiation and progression [26], in which the initiating events occur against a background of normal mutation probability and progression involves an enhanced mutation rate. In this scenario, initiation involves the mutation of genes instrumental in accurately duplicating the epigenetic patterns of genetic expression. Damage to these genes results in the failure of fidelity of vertical inheritance of gene silencing with the development of progressive clonal aberration and is associated with chromosomal instability and abnormal gene expression. These anomalies of gene expression give rise to the cytologically diagnostic features of malignancy.
The basis of this model is that carcinogenesis is viewed as taking place in two stages: (1) Initiation due to mutation of genes involved in the fidelity of vertical transmission of epigenetic pattern; and (2) Progression due to defective epigenetic inheritance by the affected clone with resultant chromatin abnormalities and inappropriate gene expression.
In the second phase it is not clear what genes have to be affected or which factors are necessary and sufficient for the expression of the malignant phenotype (such as invasion and metastasis) but, since defective epigenetic transmission will generate divergent clones exhibiting a variety of properties, it is reasonable to assume that the “effective” epimutation rate will be some orders of magnitude greater than that of the initiating mutations. An important variable influencing the epimutation rate in initiated cells is the proliferation rate of the affected population since defective vertical transmission of the epigenetic pattern is expressed only at mitosis.
Given this scenario a two-stage model of carcinogenesis similar to that proposed by Armitage & Doll [27,28] can be derived where the “instantaneous” transfer probability (p1) from a normal to a mutated pre-malignant cell is given (for small values of the mutation rate (μ)) by:
p1 = (μt)g
Where, g is the number of genes involved and t represents time. Hence, at time t, the number of cells having undergone the necessary initiating mutations outlined in the argument above is equal to the product of the integral of this transfer probability and the total size (S) of the relevant stem cell population.
In this initiated pre-malignant sub-population the probability of epigenetic errors leading to malignancy will be proportional to the proliferation rate, where k is a proportionality constant and R the mean proliferation rate.
Thus, the time-dependent probability density function of malignancy is given by:
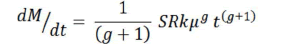
And the integral of this expression:
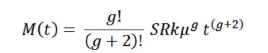
gives the cumulative incidence of cancer (M(t)) for comparison with data on the lifetime risk for different tissues [29].
Clearly there are many factors which need to be taken into account in the interpretation of this model including those relating to the number of susceptible genes (g), the relative probability (k) of the deranged pattern of gene expression giving rise to malignant behaviour, and also the mutation rate (μ) where repair rates, exposure to environmental mutagens, differences in metabolic rate and the intrinsic generation of potential mutagens such as reactive oxygen species (ROS), and the extent to which potential mutagens are removed or inactivated by metabolic pathways are modifying factors. However, making the simplifying assumption that the values of these factors are constant for different tissues, the estimated cancer risk for each tissue would be expected to be a linear function of the size of the stem cell population (S) and the mean rate of proliferation (R), a conclusion consistent with the data adduced for human cancer by Tomasetti and Vogelstein [22].
Cancer incidence in animals of differing size
An interesting extension of this argument is as follows: If cancer arises as the result of somatic mutation, and it is assumed that for similar organisms (e.g. mammals) the mutation rate for the tissues of interest falls within the same frequency range, then it can be argued from the above premises that the probability of occurrence of cancer will be related to the total number of cells of which the animal is comprised. Hence cancer incidence would be expected to vary with size and large animals should manifest a higher cancer incidence than relatively small species. However, this is not the case; apparently very large mammals, such as elephants, are paradoxically less prone to malignancy than much smaller animals [30].
Risk reduction
To some extent this surprising finding may reflect differences in metabolism although in general the cellular proliferation rates of mammals of differing size do not vary greatly and the absence of size-correlation suggests the possibility that there may be evolutionary factors that compensate for large stem cell populations. Given that it is highly probable that the normal process of copying the epigenetic pattern is subject to many possible errors and omissions, it would seem that rigorous editorial supervision is likely to feature among factors that may minimise the development of cancer and that mutations affecting this quality control mechanism would constitute an important carcinogenic lesion. Strong evidence to this effect comes from the finding that p53 mutations are present in the majority of human cancers [31] and redundant copies of this gene have been shown experimentally to greatly enhance cancer resistance in mice [32,33].
A similar conclusion may be drawn from the finding that elephants, which apparently manifest similar age-related cancer incidence to humans despite a much greater cell number, possess as many as 12 copies of the p53 gene [34]. This conclusion is consistent with a recent paper by Caulin et al. [35] in which they conclude that the available data do not support a link between body mass and the total number of tumour-suppressor genes but a weak negative correlation exists between body mass and the number of gatekeeper genes. Since gatekeeper genes are important contributors to the limitation of the mutation rate this could perhaps account for the fact that the cancer incidence for equivalent tissues are approximately the same for large and small mammals irrespective of the total cell numbers of which they are comprised. For example, in the case of colon cancer it is possible to derive incidence rates for elephants that are almost identical to those in humans by a reduction in the value for the mutation rate as illustrated in Figure 1 and Table 1. It may be, therefore, that an evolutionary feature in large animals is the development of mechanisms that diminish the mutation rate, such as the redundancy of p53 found in elephants [36].

Figure 1: Age-specific cancer incidence for colorectal cancer in humans and elephants.
Note: The actual incidence in humans (•) shown by SEER data [33] was simulated in the mathematical model using the parameter
values tabulated below and the calculated curve (blue line). The curve for the incidence in elephants (red line) was obtained using
the same values except for the size of the stem cell population (S=1.2 x 1011) and the mutation rate (μ=4 x 10-8).
| Parameters | S | R | k | µ | g |
| Human colon | 2 x 108 | 73 | 8 x 10-7 | 1 x 10-6 | 2 |
Note: S=size of the stem cell population; R=mean rate of proliferation; k=relative probability; µ=mutation rate; g=number of susceptible genes.
Table 1: The inset graph shows the derived cumulative incidence curves which are nearly identical.
References
- Holliday R, Pugh JE (1975) DNA modification mechanisms and gene activity during development. Science 187: 226-232.
- Riggs AD (1975) X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet 14: 9-25.
- Robertson KD, Keyomarsi K, Gonzales FA, Velicescu M, Jones PA (2000) Differential mRNA expression of the human DNA methyltransferases (DNMTs) 1, 3a and 3b during the G0/G1 to S phase transition in normal and tumor cells. Nucleic Acids Res 28: 2108-2113.
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251-260.
- Wigler M, Levy D, Perucho M (1981) The somatic replication of DNA methylation. Cell 24:33-40.
- Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11: 204-220.
- Jones PA, Liang G (2009) Rethinking how DNA methylation patterns are maintained. Nat Rev Genet 10: 805-811.
- Margueron R, Reinberg D (2010) Chromatin Structure and the Inheritance of Epigenetic Information. Nat Rev Genet 11: 285-296.
- Jones PA, Baylin SB (2002) The fundamental role of epigenetic events in cancer. Nature Rev Genet 3: 415-428.
- Jones PA, Baylin SB (2007) The epigenomics of cancer. Cell 128: 683-892.
- Baylin SB, Jones PA (2011) A decade of exploring the cancer epigenome: Biological and translational implications. Nature Revs Cancer 11: 726-734.
- Weisenberger DJ, Liang G (2015) Contributions of DNA methylation aberrancies in shaping the cancer epigenome. Translational Canc Res 4: 219-234.
- Riley PA (2014) Failure of fidelity of vertical transmission of epigenetic patterning as the basis of cancer. Melanoma Res 24: 424-429.
- Riley PA (2015) Cancer is the outcome of defective epigenetic copying of the pattern of selective gene activity in differentiated cells. Cancer Research Frontiers1: 280-287.
- Riley PA (2018) Epimutation and Cncer: Carcinogenesis viewed as error-prone inheritance of epigenetic information. J. Oncol
- Levene AJ, Greenbaum B (2012) Maintenance of epigenetic states by p53: The guardian of the epigenome. Oncotarget 3: 1503-1504.
- Riley PA (2017) The epigenetic theory of carcinogenesis: p53 as the guardian of the epigenome. J Oncol Cancer Res 1: 1-6.
- Leonova KI, Brodsky L, Lipchick B, Pal M, Novototskaya L, (2013) p53 Cooperates with DNA methylation and a suicide interferon response to maintain epigenetic silencing of repeats and non-coding RNAs. Proc Nat Acad Sci USA 110: E89-E98.
- Mizuno H, Spike BT, Wahl G, Levene AJ (2010) Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proc Nat Acad Sci USA 227: 22745-22750.
- Muller PAJ, Vousden KH (2013) p53 mutations in cancer. Nature Cell Biol 15: 2-8.
- Riley PA (2018) Epigenetic error and large-scale genomic instability in cancer. Biomed J Sci Tec. Res 2018: 4.
- Tomasetti C, Vogelstein B (2015) Cancer etiology: Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347: 78-81.
- Frank SA (2007) Dynamics of cancer: Incidence, inheritance, and evolution. Princeton University Press: Princeton NJ.
- Ross RK, Paganini-Hill A, Wan PC, Pike MC (2000) Effect of hormone replacement therapy on breast cancer risk: Estrogen versus estrogen plus progestin. J Natl Cancer Inst 92: 328-332.
- Pompei F, Wilson R (2001) Age distribution of cancer: The incidence turnover at old age. Hum Ecol Risk Assess 7: 1619-1650.
- Berenblum L (1974) Carcinogenesis as a biological problem. New York: Elsevier Press.
- Armitage P, Doll R (1954) The age distribution of cancer and a multi-stage theory of carcinogenesis. Brit J Cancer 8: 1-12.
- Armitage P (1985) Multistage models of carcinogenesis. Environ Health Perspectives 63: 195-201.
- Riley PA (2017) Epigenetic theory of carcinogenesis: Investigation of the model of age-specific incidence. PRAS 1: 14- 19.
- Peto R, Roe FJC, Lee PN, Levy L, Clack J (1975) Cancer and ageing in mice and men. Br J Cancer 32: 411-426.
- Hollstein M, Sidransky D, Vogelstein B, Harris C (1991) p53 Mutations in human cancers. Science 253: 49-53.
- García-Cao I, García-Cao M, Martín-Caballero J, Criado LM, Klatt P, et al. (2002) “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J 21: 6225-6235.
- https://seer.cancer.gov/
- Abegglen LM, Caulin AF, Chan A, Lee K, Robinson R, et al. (2015) Potential mechanisms for cancer resistance in elephants and comparative cellular response to DNA damage in humans. JAMA 314: 1850-1860.
- Caulin AF, Graham TA, Wang LS, Maley CC (2015) Solutions to Peto’s paradox revealed by mathematical modelling and cross-species cancer gene analysis. Phil Trans R Soc Lond B Biol Sci 370: 370.
- Caulin AF, Maley CC (2011) Peto’s paradox: Evolution’s prescription for cancer prevention. Trends Ecol Evol 26: 175-182.

Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences