Keywords
Tetracycline; Tetracycline resistance genes; Microbial community composition; Selective effect
Introduction
The intensive and extensive use of antibiotics for treating human and veterinary infections has caused antibiotic pollution, thus promoting the proliferation and accumulation of antibiotic resistance genes (ARGs) in aquatic environments [1]. The proliferation of ARGs is recognized as an emerging serious public health concern by the World Health Organization [2]. Municipal wastewater treatment plants (WTPs) continuously receive wastewater containing various antibiotic residues and ARGs. However, WTPs usually are not able to properly remove antibiotic residues and ARGs from wastewater, as revealed by the frequent detection of these contaminants in the effluent [3]. In addition, antibiotics can promote the proliferation of ARGs during the wastewater treatment process [4]. Biological wastewater treatment processes such as activated sludge can also provide suitable conditions for ARG proliferation and spread [5]. Therefore, WTPs are considered a “hotspot” for the transformation and dissemination of ARGs into aquatic environments [3,6,7].
Constructed wetlands (CWs), wetland systems are engineered for the treatment of wastewaters, are a low-cost, low-energy, easily operable and environmentally friendly wastewater treatment option. CWs can efficiently remove organic compounds (such as biochemical oxygen demand and chemical oxygen demand), total suspended solids and nutrients from wastewaters [8,9]. CWs are applied as the secondary or tertiary treatment stages for wastewater treatment process to remove nitrogen, phosphate and persistent chemicals such as phenolic compounds [10,11]. In addition, some studies have examined the potential of CWs for the removal of antibiotics and ARGs [12-14]. However, it remains unclear whether CW treatment reduces the public health risk of ARGs.
During treatment in CWs, the presence of antibiotic residues can promote the propagation of ARG-harboring bacteria, making the effluent from CWs a potential source of ARGs. Therefore, to reduce the public health risk of ARGs in the effluent-receiving aquatic environments, it is necessary to monitor the fate and distribution of ARGs in effluent from CWs. To our knowledge, however, no research has examined the public health risk of ARGs in effluent from CWs treating wastewater in the presence or absence of antibiotics.
Antibiotic residues, even at very low concentrations, can disturb microbial communities in the environment [15]. After treatment, the water flowing from a CW is a microbial source for effluentreceiving aquatic environments and the resulting change in microbial composition can affect the microbial ecology in the receiving environments.
In this study, we focused on tetracycline resistance (tet) genes. Tetracycline is an important commonly used broad-spectrum antibiotic and tet genes are often detected in wastewater effluents and variety of ecosystems [4,5,16]. Tet genes have been identified in 126 bacterial genera, both commensal and pathogenic bacteria. Because of the often acquisition in bacteria through horizontal gene transfer, tet genes are recognized as serious pollutants with public health effects [16]. The main objectives of this study were (i) to determine the effects of CWs on reducing tet gene populations from wastewater effluent in the presence or absence of tetracycline and (ii) to examine the bacterial community change via treatment of wastewater effluent by CWs. We assessed three tet genes belonging to two groups: The efflux pump genes (tetA and tetC) and the enzymatic inactivation gene (tetX). We monitored the three tet genes by real-time PCR and analyzed microbial community composition by 16S rRNA gene amplicon sequencing.
Materials and Methods
Microcosm-scale CWs and experimental design
Four microcosm-scale CWs were set up in 1 L plastic pots (100 mm diameter, 150 mm height) and operated in a greenhouse, where they were exposed to natural temperature and sunlight. Each CW was filled with 100 g of pumice rocks (grain size, 10-20 mm) from the bottom to 20 mm and with 200 g of red soil (5-10 mm) from 20 to 130 mm and was then planted with 12 to 16 common reeds (Phragmites australis). CWs were designed to treat 400 mL of wastewater in batch mode with a vertical down flow from the top to the drain at the bottom. During the 8 week start-up period, in order to construct a microbial community within each CW, they were irrigated with secondary effluent from a municipal WTP in Kofu, Yamanashi, Japan. This WTP effluent was also used as the influent wastewater for CWs in this study. The chemical properties of the effluent were as follows (mean, n=10): pH, 7.4, ammonium-N, 4.2 mg/L, nitrite-N, 0.41 mg/L, nitrate-N, 5.7 mg/L, phosphate-P, 2.2 mg/L and total organic carbon, 16.2 mg/L. During the 2 month experimental period, every 2 days CWs were filled with this wastewater, which was either injected with 230 μg/L tetracycline (hereafter “TC-contaminated CWs”) or not (“TC-uncontaminated CWs”) and drained. The effluent water from each CW collected after 2 days, 1 month and 2 months was used for downstream analysis (Figure 1).
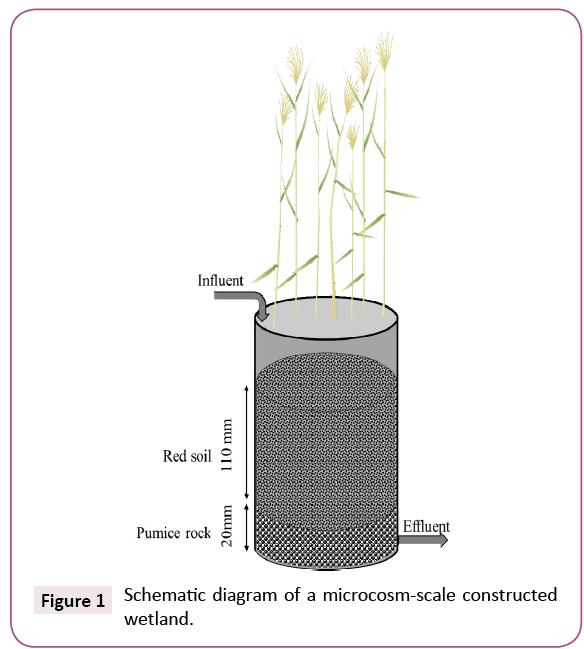
Figure 1: Schematic diagram of a microcosm-scale constructed wetland.
DNA extraction, quantitation and nextgeneration sequencing
CW effluent (about 300 mL) was collected and passed through a membrane filter (0.22 μm pore size, Millipore, Carrigtwohill, Ireland) to collect microorganisms. The filter was then subjected to DNA extraction using the NucleoSpin Soil Kit (TakaraBio, Shiga, Japan), according to the manufacturer’s instructions. The three tet genes (tetA, tetC and tetX) and 16S rRNA gene were quantified by quantitative polymerase chain reaction (qPCR) using a Thermal Cycler Dice RealTime System II (model TP900*960, TakaraBio). Each qPCR was conducted in a 25 μL reaction mixture including 12.5 μL of SYBR Green Master Mix (TakaraBio), 0.5 μL of each primer, 1 μL of template DNA and 10.5 μL of ddH2O. The qPCR temperature program was an initial denaturation at 95°C for 1 min, followed by 40 cycles of 95°C for 10 s, annealing at various temperatures for 30 s and 72°C for 20 s. After each qPCR, a melt curve analysis was conducted by increasing the temperature from 65 to 95°C to verify specificity. A standard curve for each gene was created by using a synthetic plasmid carrying the target sequence. All qPCRs were conducted in triplicate (Table 1).
| Target genes |
Primers |
Sequences |
Annealing temperature (ºC) |
References |
| 16S rRNA |
341F
534R |
5'-CCTACGGGAGGCAGCAG-3'
5'-TACCGCGGCTGCTGGCAC-3' |
60 |
Bru et al. [28] |
| tetA |
tetA-FW
tetA-RV |
5'-CCTGATTATGCCGGTGCT-3'
5'-TGGCGTAGTCGACAGCAG-3' |
61 |
Szczepanowski et al. [29] |
| tetC |
tetC-FW
tetC-RV |
5'-GCGGGATATCGTCCATTCCG-3'
5'-GCGTAGAGGATCCACAGGACG-3' |
68 |
Aminov et al. [30] |
| tetX |
tetX-FW
tetX-RV |
5'-CGTTGGACTGACTATGGCAA-3'
5'-CCCATTGGTAAGGCTAAGTCA-3' |
61 |
Szczepanowski et al. [29] |
Table 1: Target genes used for quantitative polymerase chain reaction analysis, their primer sequences and annealing temperatures.
2.3 Phylogenetic analysis of the bacterial community
Sequencing and sequence read analyses were conducted by FASMAC Corp. (Kanagawa, Japan). The extracted DNA samples from two TC-contaminated CWs and from two TC-uncontaminated CWs, respectively, were pooled together in equal ratio. These pooled DNA samples were subjected to Illumina MiSeq 16S rRNA gene sequencing. The V4 region of the 16S gene was amplified by PCR using universal primers 515F (5'-Seq A-TGT GCC AGC MGC CGC GGT AA-3') and 806R (5'-Seq B-GGA CTA CHV GGG TWT CTA AT-3') and was then read by the Illumina MiSeq Sequencer.
Sequence reads were analyzed using sickle (ver. 1.33), Fastx toolkit (ver. 0.0.13.2), FLASH (ver. 1.2.10) and USEARCH (ver. 8.0.1623_i86linux64). These analyses involved the formation of contigs, removal of error sequences and removal of chimeras. All operational taxonomic units (OTUs) were clustered at a cutoff of 0.03 (97% similarity). All OTUs belonging to archaea were removed from the data set. In addition, OTUs were eliminated from the OTU table if their relative abundance was less than 1% of the total sequences of all samples in the experiment. Alpha diversity indices (observed OTUs, Shannon, evenness and Simpson) were calculated using the QIIME pipeline [17]. In order to reduce the bias caused by different sample sizes, before microbial diversity analysis, the generated OTU table was rarified to an even sampling depth of 22074 sequences which is the minimum sequence number found in all samples. Beta diversity was determined using the UniFrac distance metric and was visualized using principal coordinates analysis [18].
Statistical analysis
The average and standard deviation (SD) of all data were calculated and all the results are expressed as mean value. Significance (P<0.05) was assessed using one-way ANOVA.
Results and Discussion
Changes in tet gene populations in wastewater with TC present
Figure 2 shows the absolute abundance of 16S rRNA and three tet genes (tetA, tetC and tetX) in influent (original wastewater) and effluent samples collected 2 days, 1 month and 2 months after the experiment started. The mean values of 16S rRNA, tetA, tetC and tetX in the influent, whole experiment time were 1.32 × 1010, 2.3 × 103, 2.9 × 102 and 4.8 × 102 copies/mL, respectively. The population stability of the three tet genes in the influent wastewater throughout the experiment confirmed that WTPs are a source for tet gene dissemination into the environment (Figure 2) [7].
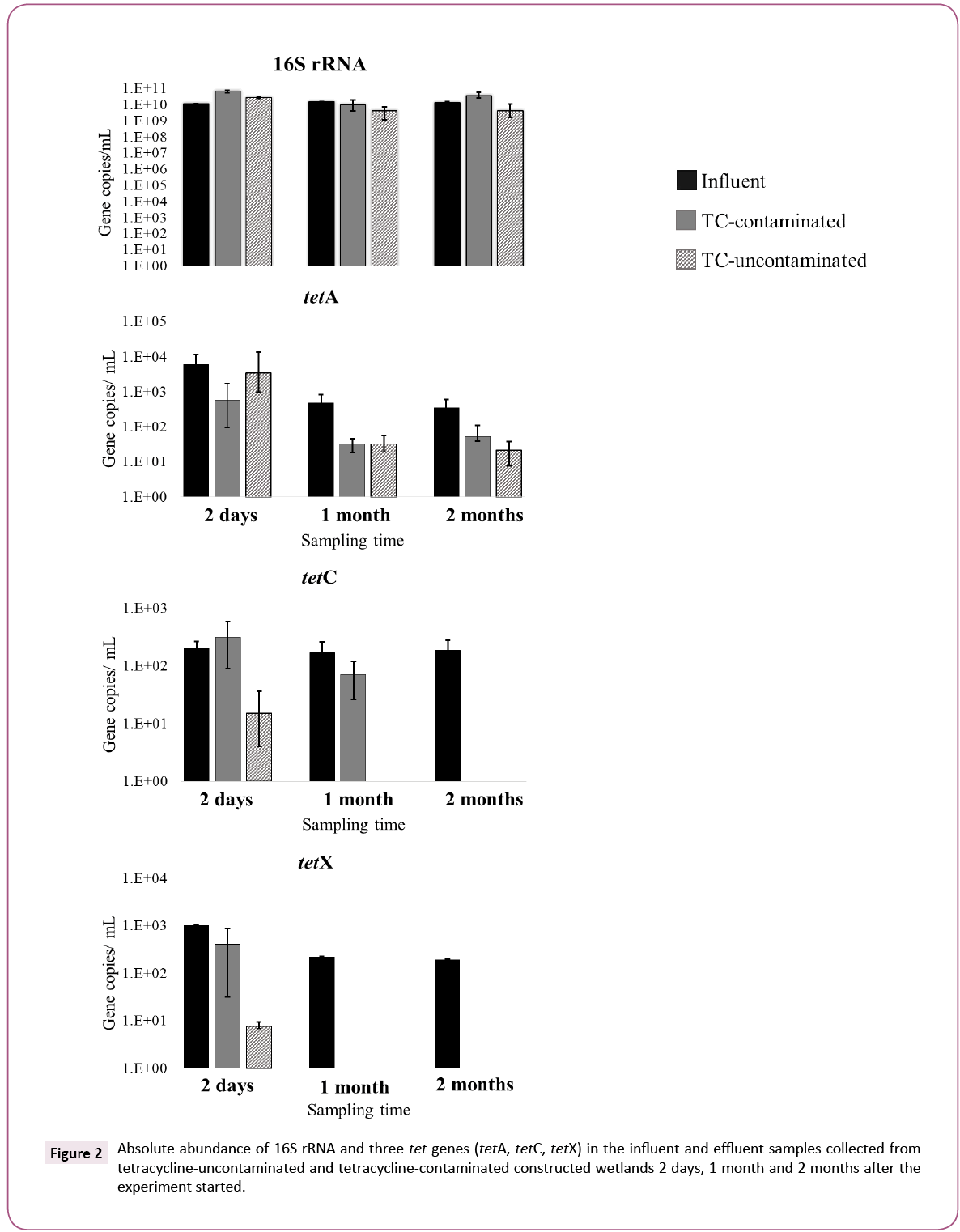
Figure 2: Absolute abundance of 16S rRNA and three tet genes (tetA, tetC, tetX) in the influent and effluent samples collected from tetracycline-uncontaminated and tetracycline-contaminated constructed wetlands 2 days, 1 month and 2 months after the experiment started.
Abundance of the 16S rRNA gene in the TC-contaminated and TCuncontaminated effluents varied between 4.2 × 109 and 3.8 × 1010 copies/mL over the 2 months. The 16S rRNA gene abundances in effluents from CWs were in range to those in the influent sample. However, tetA gene abundance in effluents collected after 1 month and 2 months from TC-contaminated and TCuncontaminated CWs were clearly lower (p<0.05) than in the influent sample. Likewise, tetC and tetX genes in effluents were also clearly lower (p<0.05) than in the influent or were below the detection limits, during 2 month experiment. A comparison between CW influent and effluent shows a high removal rate of the three tet genes in both TC-uncontaminated CWs (tetA, 2.5- 3.4 log units, tetC, 2.2-2.3 log units, tetX, 2.3-3.0 log units) and TCcontaminated CWs (tetA, 2.5-3.7 log units, tetC, 2.0-2.3 log units, tetX, 2.3-2.8 log units). These high and steady removal rates over time clearly indicate that CWs are effective at removing these tet genes and in reducing the public health risks from wastewater, whether contaminated with TC or not (Figure 2).
The abundance of the 16S rRNA gene showed temporal variation in both TC-uncontaminated and TC-contaminated effluents and no significant difference (p>0.05) was observed between the two groups, indicating that 16S rRNA gene abundance was independent of TC contamination. Berglund et al. also demonstrated that the presence of antibiotics had no effect on the abundance of the 16S rRNA gene in lake sediment microcosms [19]. In contrast, tet genes showed different trends not only between TC-uncontaminated and TC-contaminated groups but also among the three genes. In the TC-uncontaminated CWs, tetA was detected at every sampling time, but tetC was detected only in the 2 day and 1 month samples and tetX only in the 2 day sample (Figure 2).
A comparison between TC-contaminated and TC-uncontaminated effluent showed a higher abundance of tetC and tetX genes in the presence of TC. The increase of tetC and tetX in TC-contaminated effluent was also confirmed by the normalized abundance of these genes. The greater number of tetC and tetX copies in effluent of TC-contaminated CWs as compared to TC-uncontaminated CWs suggests that the presence of TC in wastewater might induce the proliferation of tetC and tetX genes during the CW treatment process. This positive selection by antibiotics on antibiotic resistance gene abundance was reported previously was a minor population in the TC-uncontaminated effluent but was the dominant one in the TC-contaminated effluent, suggesting a marked selective effect of TC on this class. The selection of Gammaproteobacteria by TC was also observed in freshwater sediment [19].
The Chlamydiae population in TC-contaminated CW increased from 5.3% to 15% after 2 months. Previous research implied that antibiotics, including TC, in aquatic environments significantly select some genera associated with opportunistic pathogens, in a manure-polluted aquatic environment and in water supply reservoirs [20,21]. On the other hand, the numbers of tetA genes in TC-contaminated and TC-uncontaminated effluents were similar after 2 months, suggesting that the presence of TC might not select for tetA. Roberts and Schwarz, reported that tetA is present in 23 genera, whereas tetC and tetX have been found in 16 and 11 genera, respectively [16]. The broader host range of tetA genes might be a reason for the different fate of tetA as compared to that of tetC and tetX, namely, the prevalence of tetA in TC-uncontaminated effluent and its independence of TC in the wastewater. These results suggest that TC-contamination in wastewater can have a selective effect on the propagation of tet genes, but the effectiveness varied among different tet genes and their hosts (Figures 2 and 3).
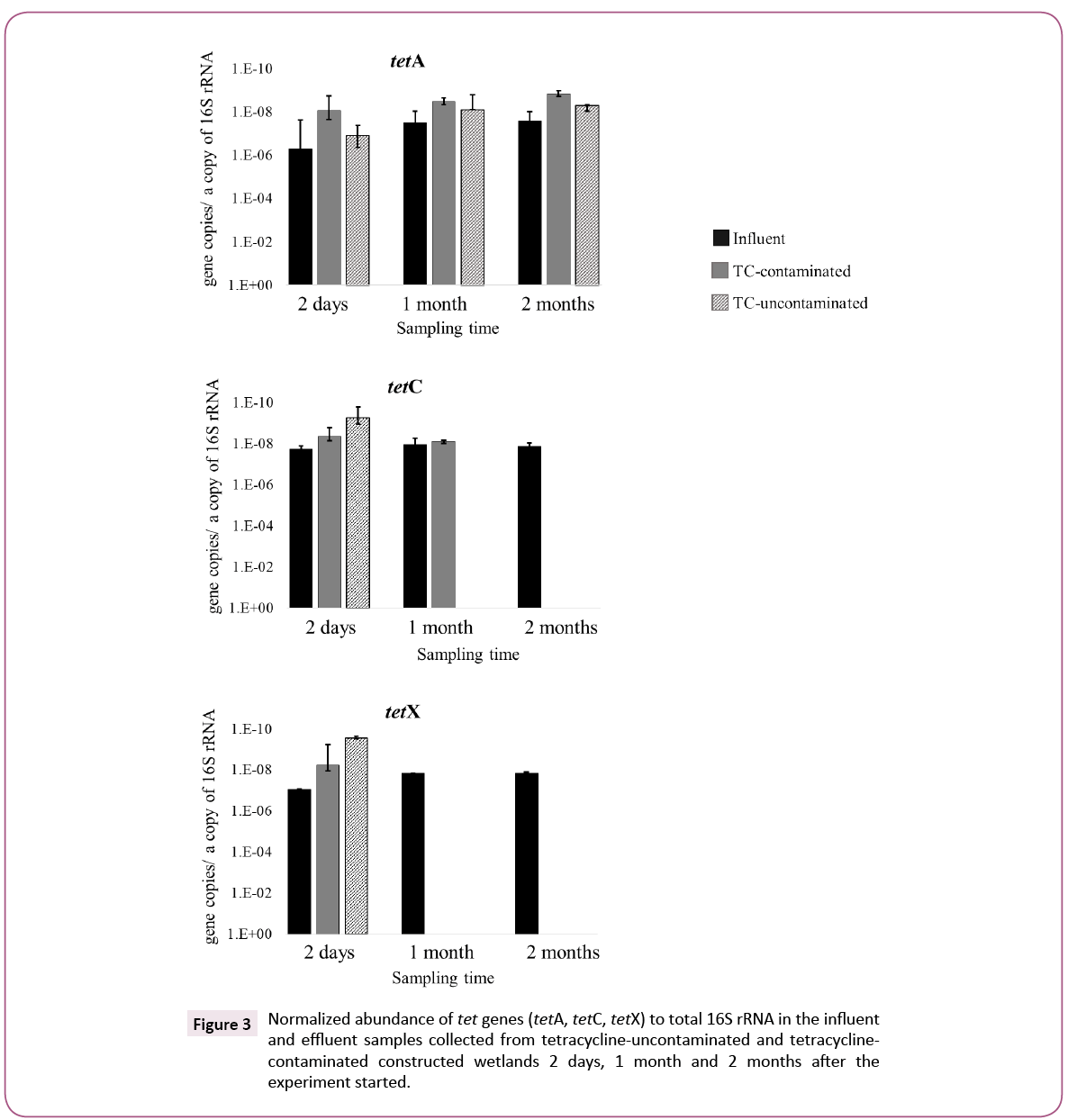
Figure 3: Normalized abundance of tet genes (tetA, tetC, tetX) to total 16S rRNA in the influent and effluent samples collected from tetracycline-uncontaminated and tetracyclinecontaminated constructed wetlands 2 days, 1 month and 2 months after the experiment started.
Changes in the bacterial community in wastewater with TC present
The bacterial communities in the influent and effluents were analyzed by comparing the V4 region of 16S rRNA gene sequences. A comparison between samples collected after same periods (e.g. 1 month, 2 months) showed that the Shannon, evenness and Simpson indices of effluents from CWs treating TCcontaminated and TC-uncontaminated wastewaters were higher than those of the influent. These results suggest that the diversity of microbial communities increased during CW treatment. The Shannon, evenness and Simpson indices in effluent from TCuncontaminated CWs were nearly constant during the 2 month experiment. These findings suggest that TC in the CWs might exert a selective effect on the population of bacterial taxa with minor abundance in CWs, thus leading to an increase in microbial diversity (Table 2).
| |
Sampling time |
Observed OTUs |
Shannon |
Evenness |
Simpson (1-D) |
| Influent |
|
134 |
4.62 |
0.65 |
0.94 |
| TC-uncontaminated |
2 days |
131 |
5.87 |
0.83 |
0.98 |
| 1 month |
155 |
5.94 |
0.82 |
0.97 |
| 2 months |
137 |
5.53 |
0.78 |
0.96 |
| TC-contaminated |
2 days |
148 |
4.81 |
0.67 |
0.91 |
| 1 month |
157 |
6.33 |
0.87 |
0.97 |
| 2 months |
141 |
5.63 |
0.79 |
0.97 |
Table 2: Alpha diversity metrics for wastewater influent and effluent samples collected from the tetracycline-uncontaminated and tetracyclinecontaminated constructed wetlands over 2 months.
A principal coordinates analysis of weighted UniFrac distance metric showed that microbial community composition in the influent wastewater was distinctively changed after treatment by either a TC-uncontaminated CW or a TC-contaminated CW. Furthermore, the analysis also revealed a large, clear separation between microbial communities depending on whether there was TC exposure or not (Figure 4).
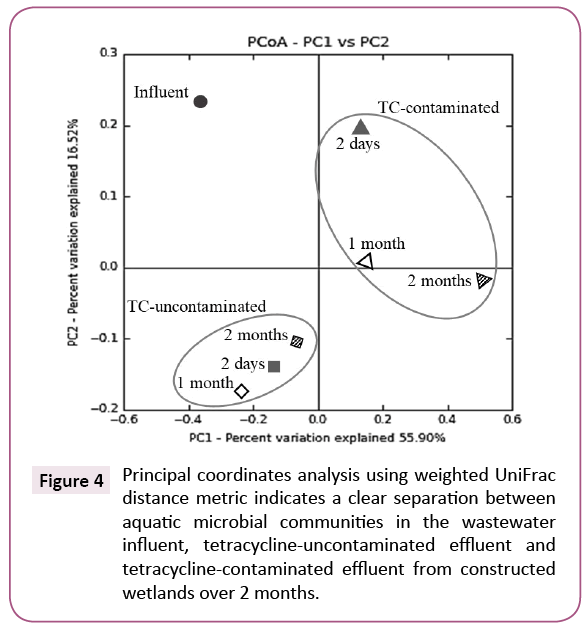
Figure 4: Principal coordinates analysis using weighted UniFrac distance metric indicates a clear separation between aquatic microbial communities in the wastewater influent, tetracycline-uncontaminated effluent and tetracycline-contaminated effluent from constructed wetlands over 2 months.
The microbial community compositions in influent and effluents from CWs after 2 months of experiment are illustrated in Figure 5. At the phylum level, microbial composition of the influent was dominated by Proteobacteria (38.9%), Actinobacteria (21.9%) and Bacteroidetes (15.5%). Proteobacteria (37.9%), Firmicutes (18.3%), Chlamydiae (15.3%) and Actinobacteria (8.4%) were dominant in effluents of CWs treating TC-uncontaminated wastewater, whereas OD1 (33.6%), Proteobacteria (25.1%), Firmicutes (8.6%) and Chlamydiae (5.3%) were dominant in those of CWs treating TC-contaminated wastewater (Figure 5).
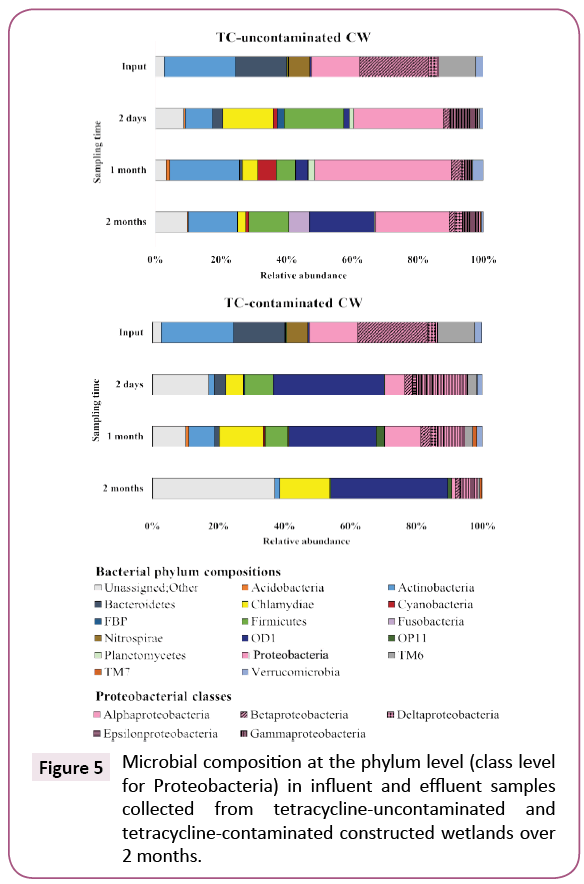
Figure 5: Microbial composition at the phylum level (class level for Proteobacteria) in influent and effluent samples collected from tetracycline-uncontaminated and tetracycline-contaminated constructed wetlands over 2 months.
The temporal variation of microbial composition was distinct between TC-uncontaminated and TC-contaminated effluents. The most changes in population were observed for the phyla Proteobacteria and Chlamydiae. Proteobacteria in the TCuncontaminated effluent (31.7-47.7%) represented a significantly larger proportion than in the TC-contaminated effluent (7.9- 25.1%). Among Proteobacteria classes, Gammaproteobacteria such as Acinetobacter, Clostridium and Salmonella [22-25]. To our knowledge, this is the first report of the selective effect of TC on Chlamydiae bacteria. Many members of this phylum are human and animal pathogens. Our results suggest that the presence of TC in wastewater might increase the public health risk of Chlamydiae in CW effluent. In addition, Cyanobacteria were found in the TC-uncontaminated CWs but not in the TCcontaminated ones, likely because TC inhibits photosynthesis in these bacteria [26]. The distinctive bacterial phyla compositions between TC-uncontaminated and TC-contaminated effluent demonstrated that TC in wastewater can select for a different microbial community. The increase of Chlamydiae and Gammaproteobacteria, two taxa that include many pathogenic bacteria, highlights the potential adverse effects on human, animal and environmental health [27].
Conclusion
This study demonstrated that CWs could remove the three tet genes (tetA, tetC and tetX) and reduce their public health risks for the effluent-receiving environment. However, the presence of TC in wastewater appears to weaken the ability of CWs to remove the tet genes. Furthermore, the presence of TC changed the microbial community composition in effluents from CWs and had a marked selective effect on the propagation of Chlamydiae and Gammaproteobacteria, which contain many pathogenic bacteria. Therefore, the removal of TC from wastewater before CW treatment or the removal of both TC and tet genes by CW will be necessary to reduce the public health risk of tet genes in aquatic environments.
References
- Baquero F, Martinez JL, Canton R (2008) Antibiotics and antibiotic resistance in water environments. Curr Opin Biotechnol 19: 260-265.
- WHO (2015) Global action plans on antimicrobial resistance.
- Zhang T (2016) Antibiotics and resistance genes in wastewater treatment plants.
- Mao D, Yu S, Rysz M, Luo Y, Yang F, et al. (2015) Prevalence and proliferation of antibiotic resistance gens in two municipal wastewater treatment plants. Water Res 85:458-466.
- Bouki C, Venieri D, Diamadopoulos E (2013) Detection and fate of antibiotic resistant bacteria in wastewater treatment plants: A review. Ecotoxicol Environ Saf 91: 1-9.
- La-Para TM, Burch TR, McNamara PJ, Tan DT, Yan M, et al. (2011) Tertiary-treated municipal wastewater is a significant point source of antibiotic resistance genes into Duluth-superior harbor. Environ Sci Technol 45: 9543-9549.
- Rizzo L, Manaia C, Merlin C, Schwartz T, Dagot C, et al. (2013) Urban wastewater treatment plants as hotspots for antibiotic resistance genes spread into the environment: A review. Sci Total Environ 447: 345-360.
- Haberl R, Grego S, Langerbraber G, Hebner A (2003) Constructed wetlands for the treatment of organic pollutants. J Soils Sediments 3: 109-124.
- Wu CY, Liu JK, Cheng SH, Surampalli DE, Chen CW, et al. (2010) Constructed wetland for water quality improvement: A case study from Taiwan. Water Sci Technol 62: 2408-2418.
- Shutes RBE (2001) Artificial wetlands and water quality improvement. Environ Int 26: 441-447.
- Vymazal J (2010) Constructed wetlands for wastewater treatment. Water 2: 530-549.
- Hijosa-Valsero M, Sidrach-Cardona R, Martin-Villacorta J, Becares E (2011) Optimization of performance assessment and design characteristics in constructed wetlands for the removal of organic matter. Chemosphere 81: 651-657.
- Anderson JC, Carlson JC, Low JE, Challis JK, Wong CS, et al. (2013) Performance of a constructed wetland in Grand Marais, Manibota, Canada: Removal of nutrients, pharmaceuticals and antibiotic resistance genes from municipal wastewater. Chem Cent J 7: 54-69.
- Nolvak H, Truu M, Tiirik K, Oopkaup K, Sildvee T, et al. (2013) Dynamics of antibiotic resistance genes and their relationships with system treatment efficiency in a horizontal subsurface flow constructed wetland. Sci Total Environ 462: 636-644.
- Halling-Sorensen B, Sengelov G, Tjornelund J (2002) Toxicity of tetracycline and tetracycline degradation products to environmentally relevant bacteria, including selected tetracycline-resistant bacteria. Arch Environ Contam Toxicol 42: 263-271.
- Roberts MC, Schwarz S (2016) Tetracycline and phenicol resistance genes and mechanisms: Importance for agriculture, the environment and humans. J Environ Qual 45: 576-592.
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335-336.
- Lozupone C, Knight R (2005) UniFrac: A new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71: 8228-8235.
- Berglund B, Khan GA, Lindberg R, Fick J, Lindgren P (2014) Abundance and dynamics of antibiotic resistance genes and integrons in lake sediment microcosms. PLoS ONE 9: e108151.
- Xiong W, Sun Y, Ding X, Wang M, Zeng Z (2015) Selective pressure of antibiotics on ARGs and bacterial communities in manure-polluted freshwater-sediment microcosms. Front Microbiol 6: 194-202.
- Huerta B, Marti E, Gros M, Lopez P, Pompeo M, et al. (2013) Exploring the links between antibiotic occurrence, antibiotic resistance and bacterial communities in water supply reservoirs. Sci Total Environ 456-457: 161-170.
- Li D, Qi R, Yang M, Zhang Y, Yu T (2011) Bacterial community characteristics under long-term antibiotic selection pressures. Water Res 45: 6063-6073.
- Barton MD (2014) Impact of antibiotic use in the swine industry. Curr Opin Microbiol 19: 9-15.
- Gomes-Neves E, Antunes P, Managero V, Gatner F, Caniça M, et al. (2014) Clinically relevant multidrug resistant Salmonella enterica in swine and meat handlers at the abattoir. Vet Microbiol 168: 229-233.
- Varela AR, Andre S, Nunes OC, Manaia CM (2014) Insights into the relationship between antimicrobial residues and bacterial populations in a hospital-urban wastewater treatment plant system. Water Res 54: 327-336.
- Gonzalet-Pleiter M, Gonzalo S, Rodea-Palomares I, Leganes F, Rosal R, et al. (2013) Toxicity of five antibiotics and their mixtures towards photosynthetic aquatic organisms: Implications for environmental risk assessment. Water Res 47: 2050-2064.
- Marti E, Variatza E, Balcazar JL (2014) The role of aquatic ecosystems as reservoirs of antibiotic resistance. Trends Microbiol 22: 36-41.
- Bru D, Martin-Lauren F, Philippot L (2008) Quantification of the detrimental effect of a single primer-template mismatch by real-time PCR using the 16S rRNA gene as an example. Appl Environ Microbiol 74: 1660-1663.
- Szczepanowski R, Linke B, Krahn I, Gartemann KH, Gutzkow T, et al. (2009) Detection of 140 clinically relevant antibiotic-resistance genes in the plasmid metagenome of wastewater treatment plant bacteria showing reduced susceptibility to selected antibiotics. Microbiol 155: 2306-2319.
- Aminov RI, Chee-Stanford JC, Garrigues N, Mehboob A, Mackie RI (2004) Detection of tetracycline resistance genes by PCR methods. Methods Mol Biol 263: 3-13.