Keywords
Melanoma, PD-1, Microparticles, T cell mimetics.
Introduction
Immunotherapy is transforming medicine and shining light on how critical immune function is in the fields of cancer, autoimmunity, and infectious disease. Checkpoint inhibitors are a powerful new class of immunotherapies. These inhibitors target co-stimulatory receptors that cancer cells use to inactivate T cells in a local tumor microenvironment. PD-1, LAG-3, TIM-3 and CTLA-4 are some of the checkpoint molecules that play critical roles in tumor-mediated immunosuppression and immune cell evasion. Checkpoint inhibition has traditionally applied antibodies to interfere with the immune checkpoint pathways to empower the anti-tumor T cell response [1,2]. Among the several checkpoint inhibitors developed, the blockade of programmed cell death protein-1 (PD-1) pathway has been approved as a treatment for patients with metastatic melanoma [3,4] PD-1 receptors expressed on T cells bind with their cognate ligands PD-L1/L2 expressed by antigen presenting cells or cancer cells. The binding of PD-1 to PD-L1/L2 can trigger inhibitory signaling downstream of T-cell receptor (TCR) and prevent the activation of T-lymphocytes [5,6]. Recent studies have also indicated that cancer cells themselves can express both PD-1 and its ligands PD-L1/L27 implicating an autocrine signaling pathway may also play a role. Kleffel et al. demonstrated the role of cancer cell-intrinsic PD-1: PD-L1 axis in promoting tumor growth independent of adaptive immunity [7].
While efficacy and patient outcomes have significantly improved, there are still concerns regarding off-target effects of blocking the PD-1/ PD-L pathway using high-affinity antibodies. Recent studies suggest that the same level of antibody blockade can inadvertently cause the over activation of T cells and severe autoimmunity in other organs such as the gastrointestinal tract where this pathway is critical to maintain tolerance [8,9]. Other risks include neurological, ocular, muscular and cardio toxicities [10]. Thus, there is an unmet need for anti- PD1 therapy to delicately modulate this pathway to prevent unwanted side-effects.
Here we report a competitive inhibitory strategy of interfering with the PD-1: PD-L1/2 interactions by using biocompatible synthetic microparticles conjugated with a soluble immune checkpoint receptor (i.e., the extracellular domain of PD- 1; (Figure 1). By conjugating PD-1 with the microparticles, we hypothesized that PD-1 will compete against the endogenous PDL-1 receptors expressed by tumor-specific T cells and melanoma cells as a soluble decoy. Thus, this approach is designed to unlock tumor-specific T cells to target tumor cells by targeting PD-1/PDL1 axis without causing downstream signaling or antibodydependent cytotoxicity. We demonstrate that both human and mouse PD-1-conjugated microparticles (PDMPs) suppress 3dimensional (3D) tumor growth of respective human A375 and mouse B16-F10 melanoma cells compared to control microparticles conjugated with the Fc portion of human IgG1 (IgG1MPs). In addition to this, in vivo studies with administration of a single local dose of PDMPs in a B16-F10 mouse melanoma model inhibited tumor growth significantly compared to control IgG1MPs at the same dose.
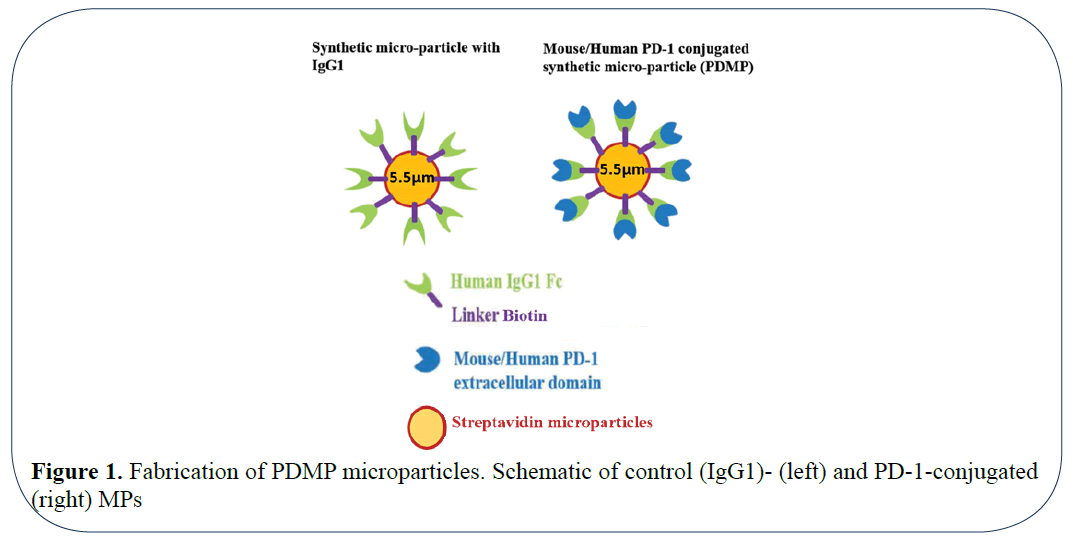
Figure 1: Fabrication of PDMP microparticles. Schematic of control (IgG1)- (left) and PD-1-conjugated (right) MPs
Materials and Methods
Melanoma Cell Lines and In vitro Culture Methods
Human melanoma A375 and mouse B16-F10 melanoma cell lines were obtained from the American Type Culture Collection (ATCC). RPMI- 1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin/ streptomycin was used to culture A375 and B16F10 mouse melanoma cells. The cells were cultured and expanded at 5% CO2 and 37ºC. For the 3-D spheroid culture, methyl cellulose (Sigma Aldrich) was used. All the cell culture reagents were obtained from Thermo Fisher Scientific, unless otherwise specified.
Microparticles, Proteins and Antibodies
Polystyrene beads coated with streptavidin of size 5.0-5.9 μm (diameter range) were purchased from Spherotech. Biotin-labeled recombinant human Fc (hFc, the Fc portion of human IgG1), human and mouse PD-1 (h/mPD-1, extracellular domain of PD-1 fused with hFc at the C-terminal) were purchased from ACRO Biosystems and BPS Bioscience, respectively. Each protein was certified with >90% purity. PE-conjugated monoclonal antibodies (mAbs) mouse anti-human PD1 (EH12.1) and hamster antimouse PD-1 (J43) (BD Biosciences) were used for flow cytometry analysis. For in vitro and in vivo PD-1 blocking studies, purified mAbs mouse antihuman PD-1 (J116) (BioXCell) and rat anti-mouse PD-1 (29F.1A12) (BioLegend) were used.
Preparation of Receptor-Conjugated Microparticles
Streptavidin coated MPs at a concentration 0.5% w/v was used for the preparation of PDMPs. The concentration of streptavidin in the MPs is approximately 1.1 × 107 moles. The MPs were centrifuged at 5000 rpm for 2 minutes. The pellet was isolated used for conjugation with respective biotin conjugated human or mouse PD-1 at room temperature for 30 minutes with gentle rotation. To validate the effect of PD-1 concentration on melanoma growth, the amount of PD-1 concentration on the microparticles was varied by altering molar folds to receptor MPs. Molar folds to receptor streptavidin coated MPs was varied between 1 × 103 to 3 × 106 (for mouse PDMPs) and 2 × 103 to 6 × 106 (for human PDMPs). The effect of concentration was further investigated by performing dilutions of these PDMPs. The control microparticles were conjugated with biotinylated human Fc (1.8 × 106 molar fold to MP). The PDMPs were stored at for storing at 4°C and used for the experiments.
Flow Cytometry Analysis
The analysis of PD-1-conjugated microparticles (PDMPs) was performed by single-color flow cytometry (Becton Dickinson). The size of unconjugated and conjugated microparticles were identified by their forward, side scatters and red fluorescence originating from PE. A minimum of 10,000 events/ sample were recorded. Data analysis was performed using the FlowJo software (Tree Star). The binding of biotinylated PD-1 to streptavidin coated MPs was confirmed using this approach. The in vitro stability of the coating was characterized using this method at different time points.
Three-Dimensional Melanoma Spheroid Culture
Melanoma tumor sphere cultures were established in above standard culture medium supplemented with 0.5% (w/v) methyl cellulose (Sigma-Aldrich). Human A375 or mouse B16F10 melanoma cells were plated at a density of 2,000 cells/well in 6-well ultralow attachment plates (Corning) and cultured using the procedure mentioned above. For determining the effect of PDMPs on the 3D spheroid formation, the cells were cultured for 9 days in the presence or absence of h/mPDMPs or control microparticles at different surface densities or anti-m/hPD-1. In addition, the effect of PDMP concentration on tumor formation was studied by varying the PDMP/ control hFc microparticle concentration in media. The media change using 0.5 mL fresh methylcellulose containing respective PDMPs or control microparticles was performed every 3 days. The results were compared to the cells treated with soluble anti m/h PD-1 (each 50 μg/ml). The spheroids were then stained with 180 μL of 0.4% (w/v) p-iodonitrotetrazolium violet solution (Sigma-Aldrich) overnight at 37°C, 5% CO2 and imaged using Nikon D3000 camera and Opteka Achromatic macro lens (10x diopter, ƒ/1.2, 18-55 mm). The number of tumor spheroids/ well in each treatment group and the controls were determined using NIH ImageJ software.
Murine Melanoma Induction in Mice
Female C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). The mice used for the experiments ranged between 7-9 weeks of age. The procedures were performed according to the guidelines provided and approved by IACUC protocol. Briefly, B16-F10 mouse melanoma cells (1 × 105 cells/inoculum) in RPMI-1640 supplemented with 0.2 mg/mL collagen were mixed with or without 5 × 104 mPDMP5.5 or hFcMP5.5 (surface saturated, conjugated with 1.8 × 106-fold IgG1). The experimental control groups were mice treated with 165 μg anti-mPD-1 or no treatment. The respective inoculum injected subcutaneously into the right flank of each recipient mouse (N=6 mice per group). Tumor formation/growth and health condition of all the mice were monitored every 2-4 days until the experimental endpoint. Mice that required euthanasia before the experimental endpoint were excluded from the study. All the other mice were sacrificed at the 15 days time point post injection. The tumors were excised, tumor mass and volume were determined. The tumor volume was calculated by the following equation [11,12]. Tumor volume (mm3) = [A (mm) × B2 (mm2)]/2, where A is the largest dimension, and B is the smallest dimension of the tumor. The harvested tumors were stored in 10% (v/v) formalin for histologic analysis.
Statistics
The results are reported as mean ± standard deviation. Statistical significance between multiple groups were calculated using One-way or Two-way ANOVA for multiple comparisons followed by Tukey post-hoc analysis (Graph pad prism). p values less than 0.05 were considered statistically significant.
Results
Characterization of Mouse and Human PD-1 Microparticles
The efficiency of microparticles coated with PD-1 would be impacted by the surface density of conjugated receptors like that of cells [13]. Another factor that would interfere with the efficacy in PDMP performance is the stability of a PD-1 coating on the microparticles. Therefore, we investigated the concentration of the m/h PD-1 coating required to saturate the PDMP surface and the stability of these coatings. This was done by selecting dilutions of purified PD-1 accounting to molar folds of 40 × 103 (low concentration of PD-1) to 3 × 106 (high concentration of PD-1) folds to the microparticles. To determine the binding of PD-1 to the microparticles, these beads were stained with PE conjugated anti-PD-1 and subsequently analyzed using flow cytometry. As shown in Figure 2, for the mouse PD-1 coated microparticles (mPDMPs), the florescent intensity of PE increased with the increase in the PD-1 concentration. The highest florescent intensity was observed in the case of 106 and 3 × 106 molar folds. Thus, this suggests that concentration of PD-1 plays a key role in coating the surface of the MPs and concentrations between 106 and 3 × 106 molar folds yield surface saturation. To address the issue of stability of the coating, this flow cytometry analysis was also performed 20 days post the coating and yielded comparable results. Since microparticle size plays critical role in modulating the efficient particle delivery to the tumor site [14,15] we chose another set of microparticles with a slightly higher particle size ranging between 6.8-7.0 μm. We studied the effect of concentration of PD-1 coating and the stability over 20 days using the same method mentioned above. The results indicate that molar fold 6 × 106 yielded the highest florescent intensity of PE after 20 days in PBS. This suggests that, for the microparticles of size 7.0 μm, surface saturation was achieved at 6 × 106 molar fold of PD-1 to the receptor microparticle.
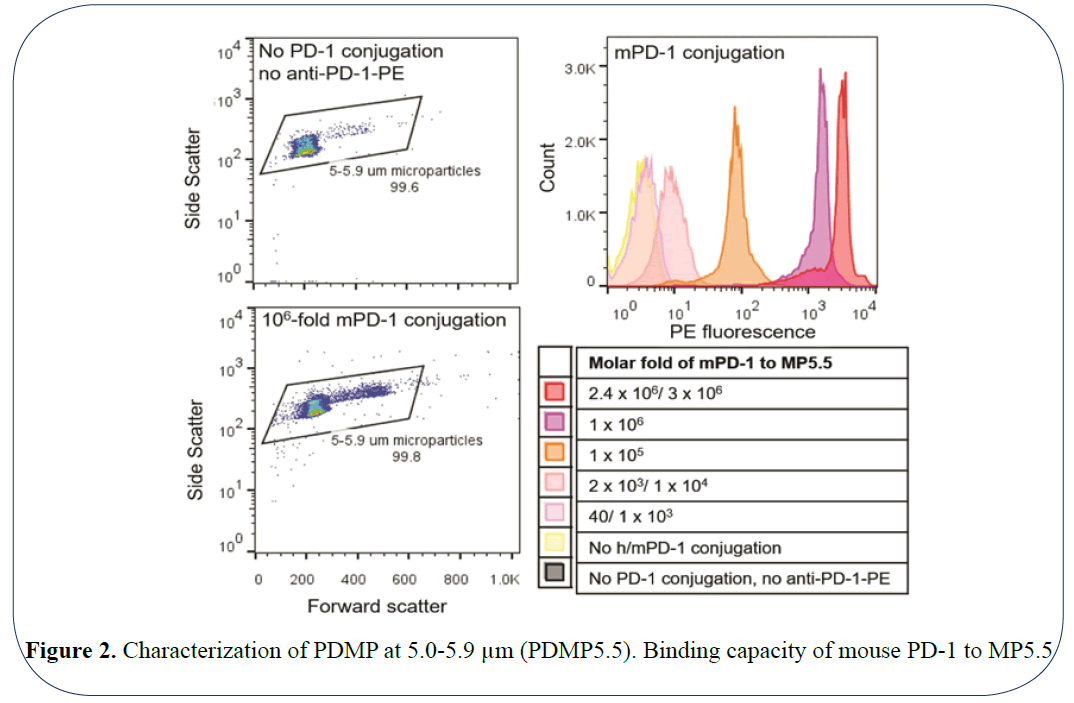
Figure 2: Characterization of PDMP at 5.0-5.9 µm (PDMP5.5). Binding capacity of mouse PD-1 to MP5.5
PDMPs Inhibit the In vitro 3D Tumor Spheroid Formation in Mouse Melanoma Cells
The ability of cancer cells to express both PD-1 and its respective ligand PDL1/2 has been reported in recent studies [7,16]. The blockade of this autocrine pathway was shown to inhibit mouse and human melanoma growth respectively, in vitro. Hence, we validated the efficacy of blocking PD1 using our microparticles on inhibiting autocrine tumor signaling in vitro using a spheroid forming assay. For this functional testing, we used mouse PDMPs of size 5.5 μm (Diameter=5.5 μm) and compared it to the groups treated control microparticles displaying IgG. As a positive control, we used a neutralizing antibody to mouse PD-1. To determine if the surface density of the PDMPs influenced tumor spheroid forming ability, we chose 3 different concentrations to 1.1 × 104/mL, 1.1 × 103/mL and 1.1 × 102/mL for 3D growth of mouse B16-F10 melanoma cells. mPDMP5.5 conjugated with 3 × 106 molar fold (100% saturated) at 1.1 × 103/mL was capable of significantly inhibiting the 3D tumor growth based on imaging (Figure 3a) and the number of spheroids/well (Figure 3b). This suggests that a highly-saturated surface density of PD-1 coating was necessary for the inhibition of tumor spheroid formation in mouse melanoma cells.
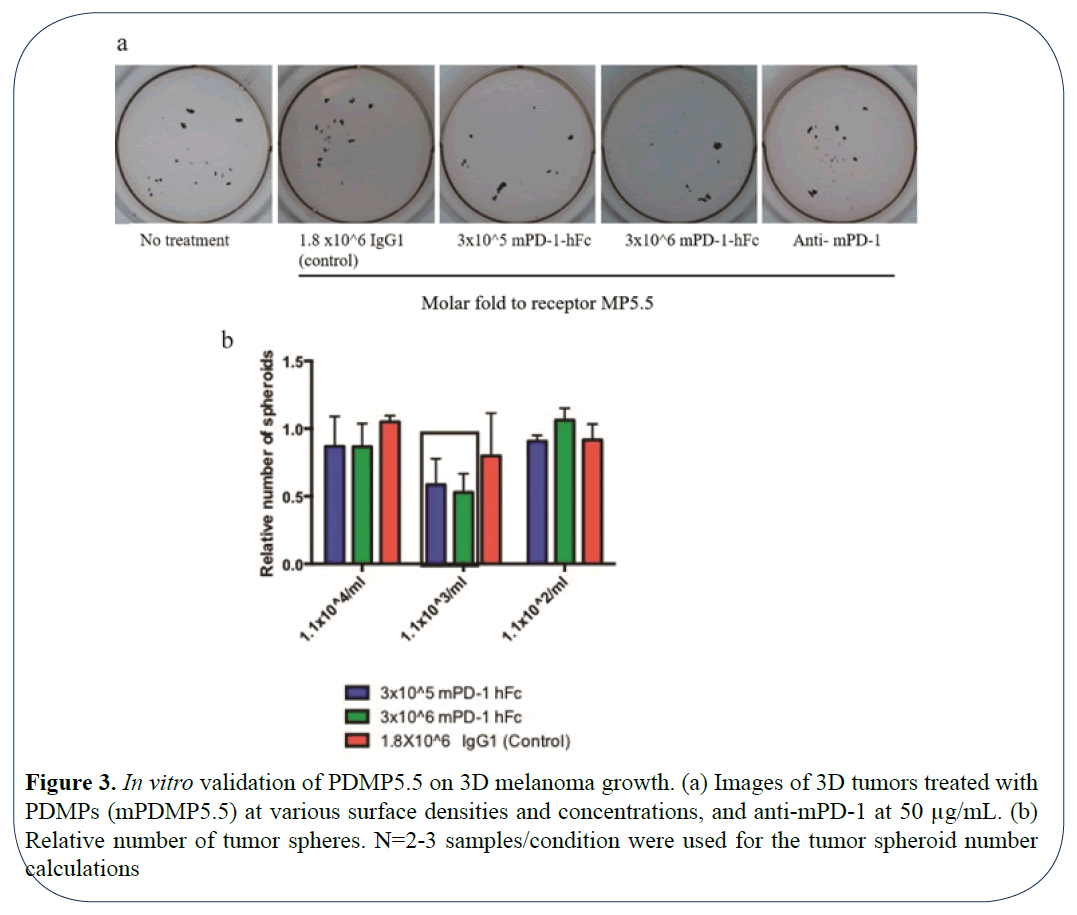
Figure 3: In vitro validation of PDMP5.5 on 3D melanoma growth. (a) Images of 3D tumors treated with PDMPs (mPDMP5.5) at various surface densities and concentrations, and anti-mPD-1 at 50 µg/mL. (b) Relative number of tumor spheres. N=2-3 samples/condition were used for the tumor spheroid number calculations
In vivo Mouse Tumor Burden Decreases with PDMP Administration
B16 melanomas are poorly immunogenic and more difficult to regress [17]. In the C57BL/6 model, it has been reported that anti-mPD-1 at 200 μg injected intraperitoneally 1 day before tumor cell inoculation and constant administration of antimPD- 1 at the same dose every 2 days post tumor cell inoculation showed slight inhibition of murine melanoma growth [7]. Similarly, in another study using anti-mPD-1 at ~20 μg locally injected onto the tumor site (when tumor sizes reached ~50 mm3), delayed melanoma growth was observed in the first few days post treatment, but the tumor relapsed [11]. Thus, we validated our mPDMPs in the same mouse melanoma model. To exclude any variations occurring due to circulation, we decided to use the respective microparticles with mouse B16-F10 melanoma cells and confine them with 0.2 mg/mL collagen for inoculation. Based on the results of in vitro 3D assays, we chose smaller MP5.5s, the optimal surface density of mPD-1 conjugated with 3 × 106 molar fold (100% saturated). Anti-mPD-1 antibody (165 μg) or microparticles with a IgG group served as experimental positive and negative controls, respectively. As demonstrated in, Figure 4a local treatment with mPDMP5.5 was able to inhibit melanoma growth remarkably at a time point of 15 days post tumor cell inoculation when compared to saline treatment or the mPD-1 treated group. The average tumor volume from the mPDMP5.5 treated group (68.3 mm3) was 4.6-fold smaller than that from the no treatment group (313.6 mm3) (Figure 4b). Similarly, the average tumor weight from the mPDMP5.5 treated group (0.13 g) was 2.6-fold lighter than that from the no treatment group (0.34 g) (Figure 4c). These results collectively indicate that PDMPs significantly reduce the tumor burden in mice. When the PDMPs were administered through intravenous injection, we observed accumulation of PDMPs in the lungs (data not shown) suggesting the need to improve the system by tuning the size of these particles for targeted tumor therapy.
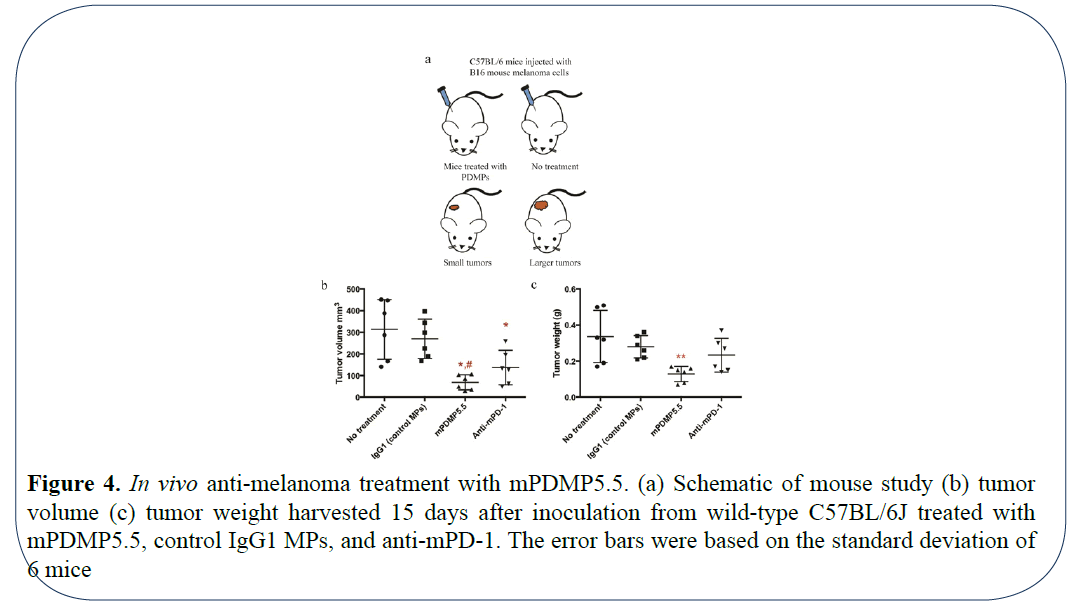
Figure 4: In vivo anti-melanoma treatment with mPDMP5.5. (a) Schematic of mouse study (b) tumor volume (c) tumor weight harvested 15 days after inoculation from wild-type C57BL/6J treated with mPDMP5.5, control IgG1 MPs, and anti-mPD-1. The error bars were based on the standard deviation of 6 mice
CD45+ Immune Cell Recruitment is Enhanced with PDMP Treatment
To determine the effect of using microparticles conjugated with PD-1 on the recruitment of immune cells in mouse, we sectioned the tissues and stained them with H&E staining and CD45 antibody. An overall increase in the CD45+ cells was observed in the tumor sections of mice treated with PDMPs and anti-PD1 when compared with IgG microparticles and no treatment groups (Figure 5). An interesting spatial localization of CD45+ cells was observed in melanoma tissues treated with PDMPs and anti PD-1. The CD45+ cells were located at the periphery of the tumor sections suggesting a possible control of the volumetric tumor growth. A recent study by Berthel et al. on the spatial localization of T cells in human colorectal cancer suggest that the presence of T cells at this invasive front leads to increased survival in patients [18]. Thus, these results demonstrate efficacy of our mPDMP5.5 in suppressing murine melanoma growth and stimulating an immune infiltration in the C57BL/6 model.
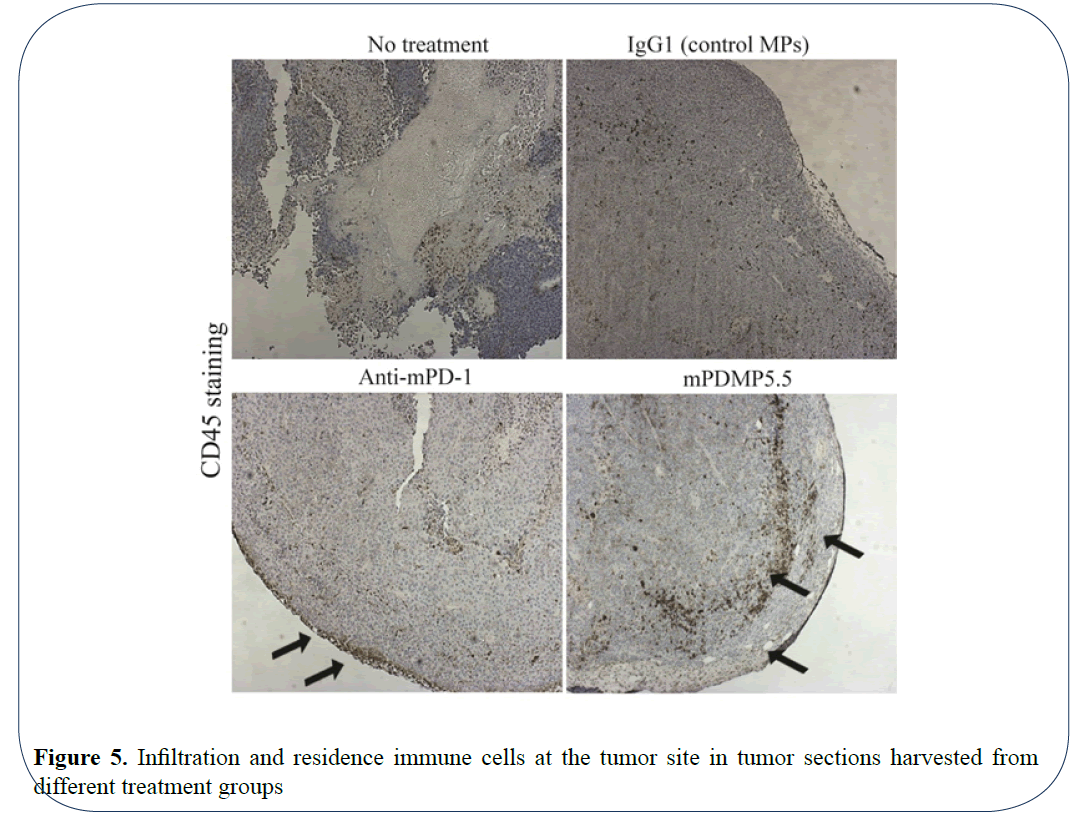
Figure 5: Infiltration and residence immune cells at the tumor site in tumor sections harvested from different treatment groups
Human PDMPs Aid in Inhibition of Tumor Sphere Formation In vitro
Following in vitro and in vivo validation of PDMPs in mouse model, we investigated the efficacy of these microparticles to target human melanoma tumor growth. Using the same method for synthesis and characterization of mPD-1 microparticles, we validated if human PDMPs could interfere with inhibition of melanoma growth in human melanoma A375 cell line. Cells treated with human PD-1 served as experimental controls. To determine if surface saturation plays a critical role on the efficiency of inhibition, we chose microparticles with coated with 2 different molar ratios of hPD-1 namely 106 molar fold (100% saturated) and 105 (~30% saturated) microparticles. In the case of hPDMPs, the 30% saturated microparticles had lesser number of tumor spheres (Figures 6a and 6b). The trend in this inhibition in sphere formation is very different from that of mouse PDMPs on B16 mouse melanoma cells, where 100% surface saturation showed the highest inhibition of tumor sphere formation. This can likely be attributed to differences between mouse and human melanoma cells. Thus, the results from our study indicate the efficacy of PDMPs to inhibit both mouse and human tumor 3D spheroid formation in vitro.
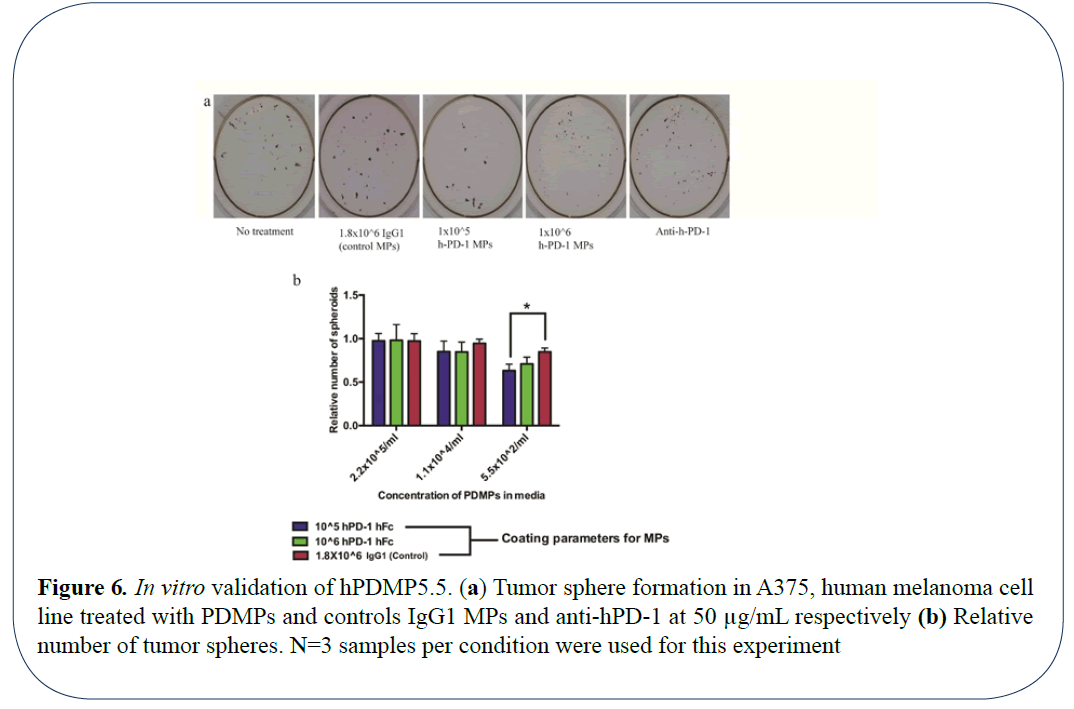
Figure 6: In vitro validation of hPDMP5.5. (a) Tumor sphere formation in A375, human melanoma cell line treated with PDMPs and controls IgG1 MPs and anti-hPD-1 at 50 µg/mL respectively (b) Relative number of tumor spheres. N=3 samples per condition were used for this experiment
Discussion
Checkpoint inhibitors are a powerful new class on immunotherapies that are actively being investigated in several other types of cancers [19,20]. Anti-PD-1 antibodies have demonstrated durable response and prolonged overall survival in a broad variety of cancer types [4,21] including advanced melanoma. The objective response rates in advanced melanoma from various clinical trials sponsored by different companies were ~ 38% [3,4,22]. Alternate approaches with soluble immune checkpoint receptors such as lymphocyte activation gene-3 protein (LAG-3), immune checkpoint receptor binding with MHC II as the ligand [23], had been developed and tested as a human IgG1 Fc fusion in phase I and II clinical trials including a monotherapy for metastatic renal cell carcinoma [24,25]. This breakthrough therapy has laid groundwork for engineering improvements to enhance a remarkable therapeutic profile.
Recent advances in the field of drug delivery have aided in building biomimetic drug carriers that are aimed to specifically target the tumor tissues and improving the outcome of drug treatment [26]. Systems such as RBC mimetic drug carriers facilitate the long-term circulation of these particles and efficient delivery to the tumor tissue [27,28]. Similarly, nanoparticles coated with neutrophil membrane have aided in targeting and capture of circulating tumor cells and efficiently decreasing tumor metastasis in vivo [29]. Integrating the advances in drug delivery to immunotherapy, in this study, we report an artificial T cell mimetic that composed of synthetic microparticles and conjugated immune checkpoint receptor PD-1 (PDMPs) to control tumor growth (Figure 7). In our system, biotin-streptavidin conjugation serves as the basis for fabrication of the PDMPs. Biotin labelled Fc binds to the streptavidin coated microparticles. The binding affinity between biotin and streptavidin is very high (Kd = 10-15 M). This aids in creating a strong linkage between Fc and a microparticle. We wanted to assure that there would be no free streptavidin molecules as they are known to be immunogenic and decrease IL2 production and activation of T cells. By saturating with high amounts of biotin, T cell activation can be restored [30]. Due to the surface saturation with biotinylated Fc and PD-1 in our system, streptavidin mediated T-cell inactivation is prevented and thereby maintain T cell function is maintained.
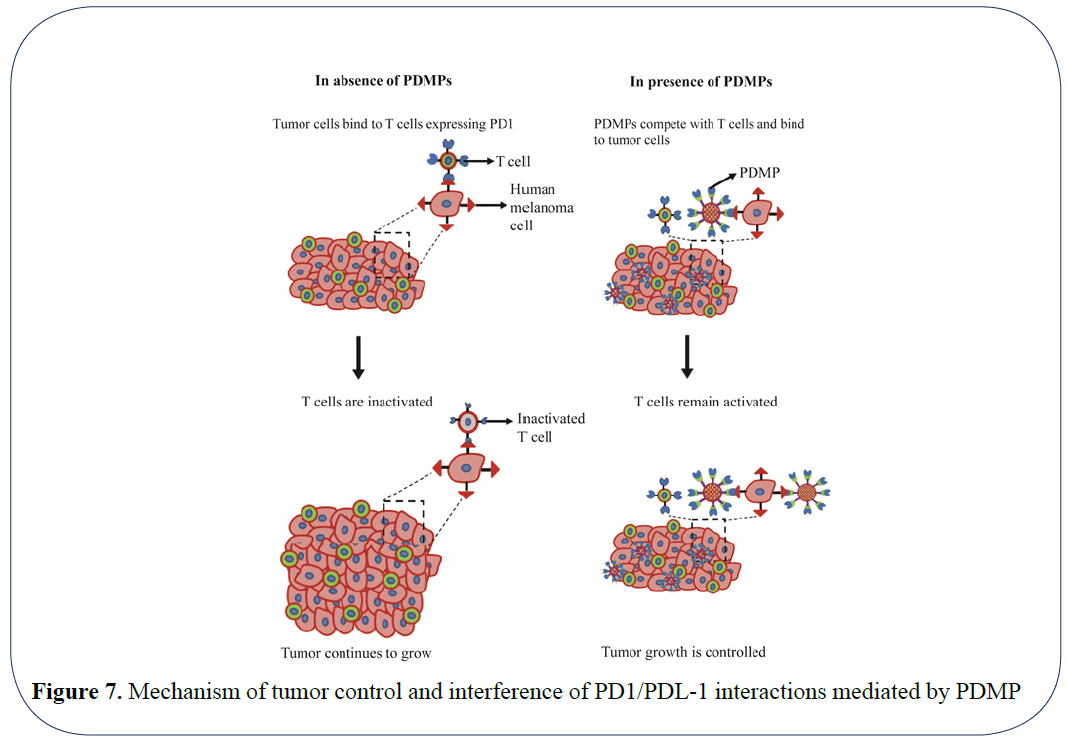
Figure 7: Mechanism of tumor control and interference of PD1/PDL-1 interactions mediated by PDMP
The PDMPs in this study suppressed human melanoma growth in vitro and mouse melanoma growth in vitro and in vivo compared to soluble anti- PD-1 blockades. This slightly improved efficacy may be attributed to the longer bioavailability of conjugated PD-1 in contrast to that of free antibody proteins [31]. The in vitro efficacy of PDMPs was validated using spheroid forming assay. To understand the molecular mechanisms of PDMP action in detail, this assay can be expanded to co-culture T-cells and cancer cells. PDMPs can be added to the co-cultures to track tumor spheroid forming ability, tumor cell viability using chromium release assay [32], activation in vitro [33]. Antibody blockade, which completely blocks immune checkpoint signaling, may cause over-activation of T cells leading to side effects, such as dose-dependent autoimmune disorders [8,34]. These mainly involve the gut, skin, endocrine glands, liver, and lung and can be severe [35]. Although steroids can be used for treating these side effects, the associated immunosuppression may compromise the antitumor response. In contrast, as partial antagonists, our PDMPs can competitively inhibit the PD-1/PDL1 signaling, potentially allowing the maintenance of the delicate inhibitory regulation of checkpoint signaling for normal immune responses.
A mouse melanoma model was used in our study investigated the administration of these microparticles intratumorally. It has been observed by others that microparticles of this size, when injected intravenously, accumulate in lungs due to large size of the microparticle [36]. Efforts to reduce lung trapping and exploiting the highly tunable nature of this particle system can significantly improve the circulation time, tissue and organ targeting, and immune cell association [37-40]. For example, low modulus (soft), discoid hydrogel microparticles have been indicated long-circulation times in blood (up to ~4 days of elimination half-life) and low accumulation in liver, kidney, lung and intestine (<10% in each organ over 5 days)40. Furthermore, the advantage of using a microparticle based system can support combination therapy [41] by having a scaffold to adhere multiple ligands. Check point inhibitors as a monotherapy and in combination with other drugs on modulating have demonstrated a synergistic increase of anti-tumor activity [42-44]. Recent studies have demonstrated the ability of soluble form of PD-1 in combination of soluble ligand of 4-1BB, a co-stimulator (member of the tumor necrosis factor (TNF)-receptor superfamily), as a gene therapy can enhance anti-tumor immunity in vitro and in vivo for murine H22 hepatocarcinoma [45]. Through engineering approaches, we can not only manipulate the type, size, shape and elasticity of particle backbones for favorable biocompatibility and bio-distribution in vivo, but also evaluate the type, combination and surface density of different receptors and ligands. The goal of future engineering and characterization will be to maintain or improve therapeutic efficacy while minimizing side effects.
Conclusion
In conclusion, this microparticle system can be designed to carry other immune check point receptors or co-stimulator ligands such as PD-1, LAG-3 etc. in combination. This will enable us to target multiple pathways involved in immune check point regulation in diverse types of tumors. Recent studies in patients demonstrate that simultaneous targeting immune check point pathways such as CTLA4 and PD-1 has improved the T cell localization and activity in tumors [46,47]. Hence, this modification can aid in improving the T cell function Thus, this system holds the potential to inhibit the intrinsic tumor growth, restore anti-tumor function of T cells and provide a “one-for-all” modality for combined immunotherapy manipulating multiple immune checkpoint pathways for higher response rates and durable and specific anti-tumor immune responses.
References
- Iwai Y, Ishida M, Tanaka Y, et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proceedings of the National Academy of Sciences. 2002;99:12293-197.
- Quezada SA, Peggs KS, Curran MA, et al. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest. 2006;116:1935-45.
- Hamid O, Caroline R, Alid D, et al. Safety and tumor responses with lambrolizumab (Anti–PD-1) in melanoma. N Engl J Med. 2013;369:134-44.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti–pd-1 antibody in cancer. N Engl J Med. 2012;366:2443-54.
- Chinai JM, Janakiram M, Chen F, et al. New immunotherapies targeting the PD-1 pathway. Trends Pharmacol Sci. 2015;36:587-95.
- Nirschl CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res. 2013;19:4917-24.
- Kleffel S, Posch C, Barthel SR, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. 2015;162:1242-56.
- Reynoso ED, Elpek KG, Francisco L, et al. Intestinal tolerance is converted to autoimmune enteritis upon PD-1 ligand blockade. J Immunol. 2009;182:2102-12.
- Wei W, Luo Z. Risk of gastrointestinal toxicities with PD-1 inhibitors in cancer patients: A meta-analysis of randomized clinical trials. Medicine. 2017;96.
- Zimmer L, Goldinger SM, Hofmann L, et al. Neurological, respiratory, musculoskeletal, cardiac and ocular side-effects of anti-PD-1 therapy. Eur J Cancer. 2016;60:210-25.
- Wang C, Yanqi Y, Gabrielle M, et al. Enhanced cancer immunotherapy by microneedle patch-assisted delivery of anti-PD1 antibody. Nano Letters. 2016;16:2334-40.
- Wang JW Improvement of the antitumor efficacy of intratumoral administration of cucurbitacin poly (lactic-co-glycolic acid) microspheres incorporated in in situ-forming sucrose acetate isobutyrate depots. J Pharm Sci. 2016;105:205-11.
- Kulpa DA, Lawani M, Cooper A, et al. PD-1 coinhibitory signals: the link between pathogenesis and protection. Seminars in Immunology. 2013.
- Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliv Rev. 2011;63:170-83.
- Win KY, Feng SS. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26:2713-22.
- Feng Z, Xiang L, Tao WH, et al. Programmed cell death 1 expression in esophageal squamous cell carcinoma and association with clinical characteristics. Indian Journal of Cancer. 2015;52:176.
- Overwijk WW, Restifo NP. B16 as a mouse model for human melanoma. Curr Protoc Immunol. 2000;39:1-29.
- Berthel A, Inka Zoernig I, Valous A, et al. Detailed resolution analysis reveals spatial T cell heterogeneity in the invasive margin of colorectal cancer liver metastases associated with improved survival. Oncoimmunology, 2017;6.
- Howie LJ, Harpreet S, Bellinda K, et al. Patient-reported outcomes in PD-1/PD-L1 inhibitor registration trials: FDA analysis of data submitted and future directions. J Clin Oncol. 2018;36:134-134.
- Hong DS, Moore KN, Patel MR, et al. Evaluation of Prexasertib, a checkpoint Kinase 1 Inhibitor, in a phase Ib study of patients with squamous cell carcinoma. Clin Cancer Res. 2018.
- Grupp SA. Advances in T cell immunotherapy for ALL. Best Pract Res Clin Haematol. 2014;27:222-8.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-30.
- He Y, Rivard CJ, Rozeboom L, et al. Lymphocyte?activation gene?3, an important immune checkpoint in cancer. Cancer Science. 2016;107:1193-1197.
- Brignone C, Escudier B, Grygar C, et al. A Phase I Pharmacokinetic and Biological Correlative Study of IMP321, a Novel MHC Class II Agonist, in patients with advanced renal cell carcinoma. Clin Cancer Res. 2009;15:6225-31.
- Fougeray S, Brignone C, Triebel F. A soluble LAG-3 protein as an immunopotentiator for therapeutic vaccines: Preclinical evaluation of IMP321. Vaccine. 2006;24:5426-33.
- Yoo JW, Irvine DJ, Discher DE, et al. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat Rev Drug Discov. 2011;10:521.
- Doshi N, Alisar SZ, Sriranjani B, et al. Red blood cell-mimicking synthetic biomaterial particles. Proceedings of the National Academy of Sciences. 2009;106:21495-21499.
- Su J, Sun H, Meng Q, et al. Long circulation red?blood?cell?mimetic nanoparticles with peptide?enhanced tumor penetration for simultaneously inhibiting growth and lung metastasis of breast cancer. Adv Funct Mater. 2016;26:1243-1252.
- Kang T, Zhu Q, Wei D, et al. Nanoparticles coated with neutrophil membranes can effectively treat cancer metastasis. ACS Nano. 2017;11:1397-1411.
- Yomogida K, Chou Y, Pang J, et al. Streptavidin suppresses T cell activation and inhibits IL-2 production and CD25 expression. Cytokine. 2012;58:431-436.
- Couvreur P, Puisieux F. Nano-and microparticles for the delivery of polypeptides and proteins. Adv Drug Deliv Rev.1993;10:141-162.
- Karimi, MA, Lee E, Michael H, et al. Measuring cytotoxicity by bioluminescence imaging outperforms the standard chromium-51 release assay. PLOS ONE. 2014;9.
- Neubert NJ, Soneson C, Barras D, et al. A well-controlled experimental system to study interactions of cytotoxic T lymphocytes with tumor cells. Front Immunol. 2016;7:326.
- Cappelli LC, Gutierrez AK, Baer AN, et al. Inflammatory arthritis and sicca syndrome induced by nivolumab and ipilimumab. Ann Rheum Dis. 2017;76:43-50.
- Michot J, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139-148.
- Decuzzi P, Godin B, Tanaka T, et al. Size and shape effects in the biodistribution of intravascularly injected particles. J Control Release. 2010;141:320-327.
- Kohane DS. Microparticles and nanoparticles for drug delivery. Biotechnol Bioeng. 2007;96:203-209.
- Champion JA, Mitragotri S. Shape induced inhibition of phagocytosis of polymer particles. Pharm Res. 2009;26:244-249.
- Merkel TJ, Jones SW, Herlihy KP, et al. Using mechanobiological mimicry of red blood cells to extend circulation times of hydrogel microparticles. Proceedings of the National Academy of Sciences. 2011;108:586-591.
- Kolhar P, Anselmo AC, Gupta V, et al. Using shape effects to target antibody-coated nanoparticles to lung and brain endothelium. Proceedings of the National Academy of Sciences, 2013;110:10753-10758.
- Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205-214.
- Larkin J, Hodi FS, Wolchok JD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New Eng J Med. 2015;373:23-34.
- Woo SR, Turnis ME, Goldberg MV, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917-927.
- Guo Z, Wang X, Cheng D, et al. PD-1 blockade and OX40 triggering synergistically protects against tumor growth in a murine model of ovarian cancer. PloS One. 2014;9.
- Xiao H, Huang B, Yuan Y, et al. Soluble PD-1 facilitates 4-1BBL–triggered antitumor immunity against murine H22 hepatocarcinoma in vivo. Clin Cancer Res. 2007;13:1823-30.
- Duraiswamy J, Kaluza KM, Freeman GJ, et al. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T cell rejection function in tumors. Cancer Res. 2013.
- Melero I, Berman DM, Aznar MA, et al. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer. 2015;15:457.