Introducción
La α-sinucleína, también conocida como SNCA es una proteína codificada por el gen SNCA. El término α-sinucleinopatía se utiliza para referirse a un grupo de enfermedades que tienen en común el depósito anormal de α-sinucleína en el citoplasma de neuronas o de células gliales [1].
La fisiopatología de estas enferemdades aún no es bien comprendida en su integridad. En la enfermedad de Parkinson (EP) y en la Demencia por Cuerpos de Lewy (DCLewy), los depósitos de α-sinucleína constituyen el componente principal de los cuerpos de Lewy y de las neuritas distróficas; también, enmenor proporción, la α-sinucleína se deposita en el citoplasma de células gliales [2]. En la Atrofia Multisistémica (AMS), la α-sinucleína se acumula en las inclusiones citoplásmicas de las células oligodendrogliales y en las neuronas, así como en las neuritas distróficas en el tronco del encéfalo [3]. Así. una diferencia anatomopatológica sustancial es que mientras en la EP y DCL el depósito de α-sinucleína es predominantmente neuronal, en la AMS lo es glial.
Una vez el cuadro clínico se ha establecido es fácil identificar los trastornos del movimiento de tipo extrapiramidal. Sin embargo muchos de estos pacientes también presentan trastornos no motores, los cuales no son de tan fácil identificación. Dentro de los trastornos no motores, en los últimos años se havenido poniendo de relieve la importancia de las alteraciones neuropsiquiátricas y neuropsicológicas [4]. El hecho de poseer una base neuropatológica común hace que resulte interesante analizar y comparar las alteraciones neuropsicológicas presentes en cada uno de estos trastornos [5]. El proceso de depósito patogénico de α–sinucleína puede iniciarse hasta varias décadas antes de que el paciente sea diagnosticado de la enfermedad. En ocasiones son los síntomas no motores, como trastorno del sueño REM, las primeras manifestaciones de la enfermedad [6].
Resulta evidente que la DCLewy presenta, “por definición”, importantes alteraciones neuropsicológicas. Quizás menos evidente, pero cada vez más conocido, es el deterioro cognitivo presente en muchos pacientes con EP y AMS. Aunque hay cierta heterogeneidad en los datos obtenidos hasta la fecha, muchos estudios recientes han demostrado que la demencia es común en la EP, y que en algunos pacientes, el deterioro cognitivo ya está presente en el momento del diagnóstico. Se ha visto que la presencia de inmunoreactividad cortical para la α-sinucleína se correlaciona con el grado de deterioro cognitivo en la EP [16]. Aarsland y cols han conducido en la última década una serie de estudios clínico-epidemiológicos que han permitido conocer con precisión la presencia de deterioro cognitivo y trastornos neuropsiquiátricos en la EP [9]. Algo similar sucede con la AMS: su conocimiento, centrado durante décadas en los trastornos motores, ha venido a considerar cada vez con mayor claridad, el deterioro cognitivo que asocian estos pacientes aún en fases tempranas de la enfermedad [7, 8].
En esta revisión se analizan los trastornos neuropsicológicos presentes en sobre cada una de estas enfermedades para finalmente comparar dichas alteraciones.
Enfermedad de Parkinson
La EP se caracteriza por ser un trastorno motor progresivo cuyos principales síntomas son la torpeza generalizada con lentitud en la realización de movimientos, escasez de motilidad espontánea, temblor de reposo y rigidez. Manifestaciones típicas son la amimia, la escasez de movimientos automáticos como el parpadeo o el braceo al caminar, la inclinación de tronco hacia delante durante la marcha, etc. Conforme aumenta la duración de la enfermedad pueden aparecer otros síntomas, como un deterioro de la y una alteración de los reflejos posturales dando lugar a caídas (Figura 1, Tabla 1). En la descripción inicial de James Parkinson ya se menciona la presencia de síntomas no motores. Así, en los últimos años, estudios clínicos y patológicos han demostrado que la EP es un proceso multisistémico caracterizado por la presencia de síntomas no motores (hiposmia, estreñimiento, depresión, trastorno de conducta del sueño REM) que acompañan o preceden al síndrome motor característico y por el depósito difuso de alfa-sinucleina, componente de los cuerpos de Lewy, que provoca la disfunción de diferentes sistemas de neurotransmisión (noradrenalina, serotonina y acetilcolina), a la vez que provoca la muerte neuronal de diferentes estructuras corticales, subcorticales y del tronco cerebral.

Figura 1: Evolución clínica de la enfermedad desde su fase presintomática, a los síntomas motores, trastornos cognitivos y complicaciones finales. Adaptado de Fahn.
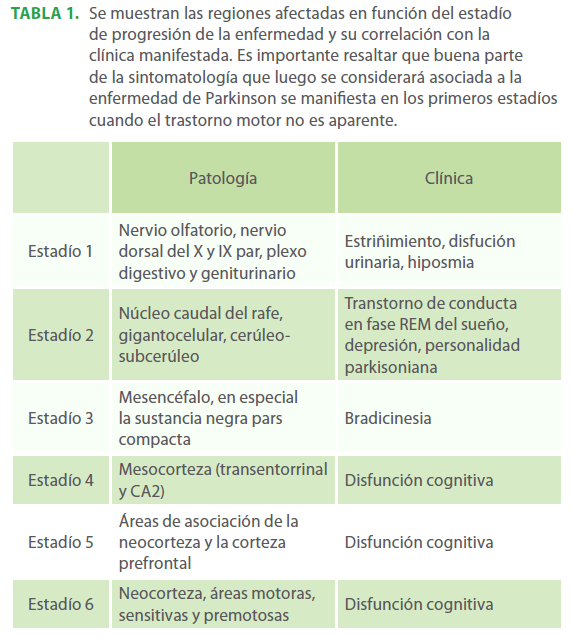
Tabla 1. Se muestran las regiones afectadas en función del estadío de progresión de la enfermedad y su correlación con la clínica manifestada. Es importante resaltar que buena parte de la sintomatología que luego se considerará asociada a la enfermedad de Parkinson se manifiesta en los primeros estadíos cuando el trastorno motor no es aparente.
Dentro de los trastornos no motores se ha visto que los factores que condicionan una mayor dependencia e insatisfacción en la EP, tanto para el paciente como para sus familiares, son junto con la clínica motora con pobre respuesta a la terapia dopaminérgica (disartria, disfagia y alteración de los reflejos posturales), los trastornos neuropsicológicos y los trastornos neuropsiquiátricos.
El deterioro cognitivo y los trastornos neuropsiquiátricos están presentes en muchos pacientes con EP. Aunque hay cierta heterogeneidad en los datos obtenidos hasta la fecha, muchos estudios recientes han demostrado que la demencia es común en la EP, y que en algunos pacientes, el deterioro cognitivo ya está presente en el momento del diagnóstico. Aarsland y cols. han conducido en la última década una serie de estudios clínico-epidemiológicos que han permitido conocer con precisión la presencia de deterioro cognitivo y trastornos neuropsiquiátricos en la EP [9]. Cuando se habla de alteraciones neuropsicológicas en la EP resulta crucial distinguir cuándo se habla de la EP sin demencia y cuándo de la Demencia asociada a la Enfermedad de Parkinson (DEP).
La prevalencia puntual de la DEP es cercana al 30% y la prevalencia acumulada es muy alta: al menos el 75% de los pacientes con EP que sobreviven más de 10 años desarrollan demencia. El tiempo medio desde el inicio de la EP hasta la aparición de la demencia es de aproximadamente 10 años (9). Sin embargo, hay variaciones considerables. Utilizando una amplia gama de medidas neuropsicológicas se identificaron tres subgrupos de pacientes con EP en comparación con los controles: un subgrupo PD no mostraron deterioro cognitivo o mínimo, un segundo grupo mostró un patrón variable o incierto de deterioro cognitivo leve (MCI) a grave, y un tercer grupo donde había evidencia de deterioro cognitivo grave en la mayoría de los dominios cognitivos [10-13]. Entre los pacientes PD-MCI, casi dos tercios tenían subtipo de MCI no-amnésico, y un tercio, subtipo de MCI amnésico. Recientemente se ha demostrado que las bases neuroanatómicas subyacentes en ambos tipos de MCI son distintas: la densidad de la sustancia gris en las cortezas temporal derecha y cingular posterior es menor en los pacientes con MCI no amnéstico, mientras que la densidad de sustancia gris en pacientes con MCI amnéstico era inferior en el precúneo, corteza prefrontal izquierda y área motora primaria [14].
La aparición de deterioro cognitivo en pacientes con EP se ha detectado incluso en las primeras etapas de la enfermedad. Por tanto, el estado cognitivo en la EP se debería investigar de forma rutinaria, ya que la detección precoz del deterioro cognitivo es crucial para un manejo preciso y un tratamiento oportuno. A continuación se analizan estas funciones individualmente:
La atención elemental aunque está claramente alterada en la DEP está conservada en los pacientes con EP sin demencia, si bien la puntuación en diversas tareas que requieren un mantenimiento voluntario o la resistencia a la interferencia suele encontrarse algo disminuida respecto a controles sanos [20].
Muchas investigaciones han observado íntima conexión de los ganglios basales a la parte inferior de los lóbulos frontales y la disminución de salidas dopaminérgicas a estas regiones en casos de pacientes con Parkinson. Estas investigaciones han sugerido que los déficits cognoscitivos en la EP no reflejan problemas en el funcionamiento de los ganglios basales sino una desconexión a los lóbulos frontales. Se postula que las desconexiones fronto-talámicas explican la pérdida de espontaneidad e imaginación y la falta de iniciativa que presentan los pacientes con EP [17]. En un cuestionario de funciones cotidianas los pacientes con EP fueron clasificados diferentes a los normales en conductas relacionadas a la iniciativa, conductas estereotipadas, indiferencia, desinterés, dependencia social, y control intelectual. El enlentecimiento para dar respuestas y la tendencia a verbalizar pero no ejecutar movimientos correctos puede también dar evidencia del involucramiento de los lóbulos frontales en la EP [18].
Evidencias experimentales con monos en los que se utilizaron tareas de respuestas demoradas [19] sugieren que las lesiones en los ganglios basales producen síntomas muy parecidos a los que se observan después de la ablación bifrontal cortical. La estimulación eléctrica del caudado también ocasiona alteraciones en la ejecución de respuestas demoradas en monos. Los pacientes con EP también han demostrado dificultad en este tipo de tareas, aunque ésto puede presentarse en aquéllos pacientes con déficits cognoscitivos severos. En estas tareas de respuesta demorada se observó que aquéllos pacientes con EP que habían padecido talamotomía tenían más dificultades en tareas de apareamiento visual que los sujetos normales. Los déficits en estas clases de tareas están asociados con patología en las proyecciones principales de los núcleos dorsomediales del tálamo a los sistemas frontales dorsolaterales y orbitales [12,16].
La afectación de la memoria es moderada. Existe un déficit de evocación con almacenamiento relativamente conservado. A pesar de la diversidad y heterogeneidad de los hallazgos, parece claro que la memoria visual es la más frecuentemente disminuida y que la memoria episódica a largo plazo no se altera habitualmente [20].
Diversos estudios han reportado alteraciones en la memoria en los pacientes con EP aún cuando, existe controversia respecto a esto, sobre todo debido a las pruebas utilizadas, a la población estudiada y también a las diversas hipótesis explicando las razones por las cuales los pacientes presentan alteraciones en la memoria. Algunos autores han estudiado diferentes procesos específicos de los trastornos de memoria. Así, los parkinsonianos inicialmente pueden codificar la información adecuadamente, pero la organización y la consolidación requieren de más tiempo para que se activen estrategias de búsqueda, por lo que los pacientes, aparentemente presentan una ejecución adecuada en tareas de memoria, particularmente si tienen el tiempo suficiente para consolidar la información [13,16].
En general, los pacientes con EP puntúan dentro de los límites normales en las pruebas que involucran procesos automáticos, reconocimiento inmediato, acceso a la memoria semántica a largo plazo, rastreo de memoria a corto plazo. Sin embargo, los pacientes con EP han demostrado dificultades en tareas que requieren reconocimiento tardío, evocación inmediata o tardía. Estos resultados apoyan la idea que los déficits de memoria en EP son aparentes únicamente cuando la tarea requiere de la integración de información de diferentes sistemas de almacenamiento de memoria y no cuando se requiere del almacenamiento y evocación de información per sé [11,20].
Los trastornos visuoespaciales se encuentran frecuentemente en pacientes con EP. Existen dificultades en apreciar la posición relativa de los objetos en el espacio y en integrarlos de forma coherente, así como la ejecución de operaciones mentales que implican conceptos espaciales. Se asocia a una progresión del déficit cognitivo y alteraciones de la marcha [10,15].
Existen diversos estudios que sugieren la presencia de déficits visuoespaciales en pacientes con E.P. Algunos investigadores afirman que posiblemente la función más afectada en pacientes con EP se refiere a los procesos visuoespaciales. Sin embargo, en este tipo de evaluaciones es importante aclarar que la exploración de funciones visuoespaciales es muy heterogénea ya que involucra diversos aspectos: exploración espacial, discriminación visual, orientación espacial, percepción de ángulos, memoria topográfica, percepción espacial, y habilidades construccionales entre otras. La alteración básica se debe a una dificultad en la orientación espacial y visuopostural las cuales dependen de las conexiones entre los ganglios basales y la corteza frontal, sugiriendo que el patrón de deficiencias se parecen a las alteraciones que presentan pacientes con lesiones en el hemisferio derecho. Después de varios estudios en este campo se concluyó que los pacientes con EP con síntomas predominantes en el lado izquierdo pueden diferir comportamentalmente de aquéllos con síntomas predominantes en el lado derecho y que los pacientes pueden tener déficits específicos en cuanto a mantener la orientación espacial, particularmente cuando se requiere que se haga un cambio en la orientación [12,16].
Los resultados de estos estudios sugieren que los pacientes con EP tienen un déficit visuoespacial tanto en tareas que requieren respuestas motoras como las que no. Aún no está clarosi existe un déficit específico en orientación espacial mediado por los ganglios basales, o se debe a un déficit para cambiar de set mental, que, tal vez refleja una disminución en la estimulación de los ganglios basales a la corteza frontal. Este interrogante no puede ser fácilmente aclarado ya que muchos de los estudios utilizan tareas que miden varias funciones y no describen cuidadosamente la población en términos de variables como severidad de la enfermedad y síntomas motores del lado predominante [11,15].
Respecto al lenguaje se describen déficits moderados en tareas de nominación, pero no la presencia de trastornos graves de la comprensión ni anormalidades parafásicas características de las demencias corticales [21]. La mayoría de los enfermos parkinsonianos sufre una disartria hipocinética. La fluidez verbal está afectada y existe un enlentecimiento en el proceso de generación del lenguaje, especialmente en la denominación de acciones [22]. También se han descrito déficits de lectura y escritura relacionados con las incapacidades motoras. En muchos pacientes aparece una alteración de la escritura, también hipocinética, con presencia de micrografía.
En estudios neuropsicológicos casi no se reproducen problemas de lenguaje en pacientes con EP, aparentemente se encuentran intactos en aspectos lógico-gramaticales y lógico- verbales complejos, pero tienen dificultades para realizar análisis detallados ó estrategias adecuadas para solucionar y verificar problemas. La enfermedad involucra básicamente el componente motor tanto en sus aspectos articulatorios, la velocidad y el aspecto entonacional como el volumen del lenguaje. No se observan alteraciones en los diferentes niveles integrativos del lenguaje, por lo que las alteraciones motoras, incluyendo la reducción y la lentificación están vinculadas al sistema de ejecución a nivel subcortical [10-13].
En cuanto a las praxias, un estudio de pacientes con EP, más del 60% tenía un examen de las praxias peor que los sujetos control, siendo atribuido por la mayoría de los autores a disfunción frontal [20].
A pesar de las evidencias en favor de la importancia de los déficits dopaminérgicos en la génesis del deterioro cognitivo asociado a la EP, no hay consenso acerca del efecto clínico de la levodopa (LD) sobre el deterioro cognitivo. De hecho, se ha comunicado tanto que la levodopa mejora como que no afecta el rendimiento cognitivo frontal y/o las funciones de memoria [23]. La respuesta a estas variaciones en el efecto de la LD parece explicarse por la alta heterogeneidad de las muestras entre estudios. Otros autores encontraron un efecto deletéreo de la LD sobre ciertos dominios cognitivos al estudiar muestras de pacientes fluctuantes o en fases avanzadas de la enfermedad, mientras que los estudios con una clara mejoría cognitiva son aquellos que han estudiado muestras con pacientes en fases más precoces. Bajo esta perspectiva, los estudios de Kulisevsky se han basado en una correcta homogeneización de las muestras, evaluando el efecto diferencial de la LD o con respuesta fluctuante. Esto ha permitido detectar que la LD mejora tanto las funciones ejecutivas como la memoria en los pacientes de novo, aunque sin llegar a un rendimiento normal no modifica la función cognitiva en los pacientes con una respuesta estable y empeora tanto las funciones ejecutivas como la memoria en pacientes fluctuantes al administrar el fármaco, volviendo a los valores previos a las 4 horas. Además y después de un seguimiento de pacientes de novo en tratamiento con levodopa durante 2 años, se observaba que los beneficios iniciales observados en los pacientes estables se pierden de manera progresiva a partir de los 12 meses [24,25].
Por tanto el reemplazo dopaminérgico central produce sólo una mejoría incompleta de la función cognitiva de estos pacientes, ya que sólo mejora algunas de las tareas cognitivas alteradas y porque la mejora inicial se estabiliza en los primeros años para, posterioremente, seguir deteriorándose según avanza la enfermedad y aparecen las fluctuaciones motoras. Una de las explicaciones por las que el reemplazo dopaminérgico no compensa todos los déficits cognitivos es que en su deterioro intervienen otros sistemas de neurotransmisión. En la EP existe una pérdida de neuronas noradrenérgicas, serotoninérgicas y colinérgicas. De entre estos sistemas de neurotransmisión deficitarios de la EP, las alteraciones de las vías de proyección colinérgicas son las que se han relacionado de manera más consistente con el deterioro cognitivo de los pacientes [26].
Demencia con Cuerpos de Lewy
La demencia con cuerpos de Lewy (DCLewy) se caracteriza por parkinsonismo de intensidad variable, rasgos psicóticos como las alucinaciones visuales (AV), y fluctuaciones del estado cognitivo que afectan especialmente a la atención y concentración. La DCLewy podría constituir del 10% al 36% de las demencias degenerativas, sólo sobrepasada por la EA, a la que se puede asociar. La edad de comienzo de la DCLewy suele ser entre los 70 y los 80 años, como ocurre con otras demencias degenerativas [27].
El hallazgo histológico definitorio de la DCLewy es la presencia en las neuronas corticales de los cuerpos de Lewy, inclusiones citoplasmáticas eosinófilas y redondeadas cuyo principal componente es la α-sinucleína, además de ubiquitina y proteínas de neurofilamento. Aparecen también abundantes placas de amiloide (como las de la EA), y escasos ovillos neurofibrilares [31].
La DCLewy es diagnosticada cuando la demencia precede al parkinsonismo o aparece al mismo tiempo que este. La demencia de la enfermedad de Parkinson debería utilizarse para describir la demencia que aparece en el contexto de una enfermedad de Parkinson ya bien establecida. En el caso de los estudios de investigación que deban distinguir entre DCLewy y demencia de la EP, se recomienda que para diagnosticar DCLewy se use como norma que la demencia no debería haber comenzado más allá de un año tras el inicio del parkinsonismo [21].
Son también frecuentes la atención fluctuante, la lentitud del pensamiento, la hipersomnia diurna, y los estados de ausencia. A menudo el insight está preservado, a diferencia de los casos de EA. Las fluctuaciones del rendimiento cognitivo se presentan de un día para otro o incluso en el mismo día, y son frecuentes los cuadros confusionales de origen desconocido, no atribuibles a factores externos ni médicos definidos [42].
Los signos parkinsonianos son, quizá, el rasgo más característico de la DCLewy. En algunos casos preceden a la demencia, de forma sutil o notoria, y los pacientes son diagnosticados durante meses o años de EP, con respuesta escasa y transitoria a la L-Dopa. Predomina la rigidez, que aparece precozmente en más de la mitad de los casos, pero son también frecuentes la bradicinesia, la falta de expresividad facial, los trastornos de la postura, marcha y reflejos posturales y, en menor medida, la hipofonía y el temblor.
Desde el punto de vista motor las diferencias entre el parkinsonismo de la DCLewy y de la EP son escasas: el carácter simétrico, escaso temblor y poca respuesta al tratamiento con L-Dopa. Se aconseja no utilizar el diagnóstico de DCLewy cuando el parkinsonismo precede a la demencia en más de un año, calificándose estos casos de DEP.
La mezcla de características clínicas y su orden de aparición en el curso de la enfermedad pueden variar de paciente a paciente, dependiendo de dónde estén localizadas predominantemente las lesiones histopatológicas. Por ejemplo, los pacientes con signos extrapiramidales precoces y prominentes muestran marcados cambios nigroestriatales, mientras que aquellos con alucinaciones y deterioro cognitivo tempranos tienen una pronunciada participación cortical o del sistema límbico. De la misma manera, los pacientes con DCLewy con inestabilidad postural y caídas muy frecuentes presentan lesiones histopatológicas de la enfermedad en médula espinal y ganglios simpáticos y parasimpáticos.
El rasgo psicótico más claro es la presencia de alucinaciones visuales, típicamente complejas y muy elaboradas, y de presentación precoz. A veces las alucinaciones se acompañan de ideas delirantes, especialmente del tipo persecutorio, muy establecidos, elaborados y de fuerte contenido, a diferencia de los pobremente formados de la EA [32].
En comparación con controles sanos, la función cognitiva de los pacientes con DCLewy está deteriorada en todas sus áreas, con gran variabilidad. Los pacientes con DCLewy muestran un deterioro de las capacidades visuoperceptiva, visuoespacial, atencional y ejecutiva [29]. Estos hallazgos son congruentes con la distribución prototípica de Cuerpos de Lewy en la corteza frontal, cingular y temporal posterior [28,35].
Los defectos en la percepción visual (distorsiones de tamaño, forma, movimiento o color), en combinación con defectos generales como escasa luz externa, confusión y deterioro cognitivo, juegan un papel clave en el desarrollo de las alucinaciones visuales, las identificaciones erróneas delirantes, las agnosias visuales y las incapacidades visuoconstructivas características de la DCLewy [31,33].
A pesar de que la memoria se encuentra globalmente menos afectada que en otras enfermedades neurodegenerativas como la EA [29], los pacientes con DCLewy (al igual que los que padecen EA) sufren una clara afectación de la memoria semántica (no así de la memoria episódica). Los pacientes con DCLewy en particular tienen problemas cuando tienen que extraer el significado de imágenes, lo que muy probablemente se debe a una combinación de deterioros semántico y visuoperceptivo [30,34].
Atrofia Multisistema
La Atrofia Multisistémica (AMS) es una enfermedad neurodegenerativa que ha sido descrita durante décadas con diferentes nombres de acuerdo a las características clínicas predominantes; así, la atrofia olivopontocerebelosa (OPCA), por tener primordialmente manifestaciones cerebelosas, fue descrita por primera vez por Dejerine y Thomas en el año 1900. La forma con predominio de disautonomía fue reportada por Shy y Drager en 1960; finalmente la que cursa con características parkinsonianas, degeneración estrionígrica, fue comunicada por van der Eecken en 1960 y por Adams en 1964 [36,37]. Pero fue en el año 1969 cuando se introduce el término AMS, para denominar a los cuadros neurodegenerativos progresivos que ocurren esporádicamente y se caracterizan por parkinsonismo, disfunción cerebelosa e insuficiencia autonómica en diferentes proporciones (38). Según estudios epidemiológicos la prevalencia es de 1.94-4.5 por 100000 habitantes y con una incidencia promedio de 3 nuevos casos por 100 000/año [43,44].
El cuadro parkinsoniano incluye a la bradicinesia con rigidez, inestabilidad postural, temblor, pobre respuesta a levodopa o buena respuesta pero solo al inicio. La disfunción cerebelosa consiste en ataxia de la marcha y de extremidades, trastorno del lenguaje y de los movimientos extraoculares. La insuficiencia autonómica se manifiesta como hipotensión ortostática, disfunción eréctil, constipación y disminución de la sudoración; así también, incontinencia urinaria y nicturia [39]. Sin embargo, actualmente se simplifica en dos subtipos clínicos: AMS-P y AMS-C de acuerdo a si el trastorno motor es predominantemente parkinsoniano o cerebeloso [40] (Figura 2). Este trastorno afecta a ambos sexos, inicia frecuentemente en la edad adulta y tiene un promedio de supervivencia de 9 años desde la aparición del primer síntoma [41].
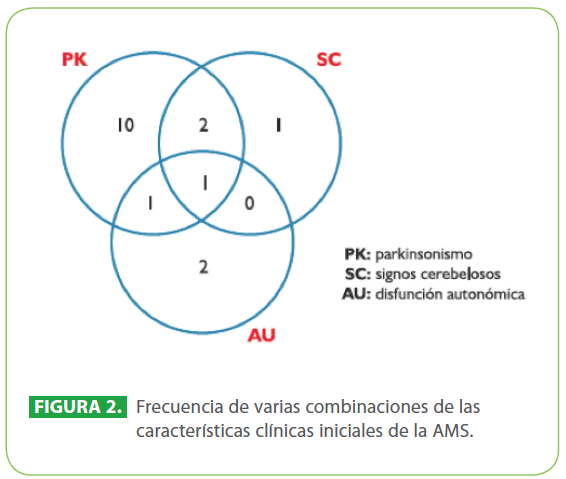
Figura 2: Frecuencia de varias combinaciones de las características clínicas iniciales de la AMS.
De etiología incierta se caracteriza a la histopatología por la presencia de inclusiones citoplasmáticas gliales [2,43], y hace algunos años se descubrió mediante técnicas de inmunotinción a la α-sinucleína como marcador patológico de esta entidad por lo que actualmente se la clasifica como una α-sinucleinopatía [2]. Los tests de función autonómica y neuroimágenes no son parte de los criterios diagnósticos pero frecuentemente son utilizados para sustentar el diagnóstico y excluir otras condiciones. Un anillo hiperintenso en el borde lateral del putamen ha sido reportado en alrededor de un tercio de pacientes con AMS en el estudio de Wenning et al. [41]. Sin embargo, la resonancia magnética puede ser normal hasta en el 20% de los casos [45]. Los tests de función autonómica pueden ayudar a corroborar los signos y síntomas del paciente o a detectarlos cuando son asintomáticos mas no puede diferenciar entre AMS y otras enfermedades que también cursan con trastornos autonómicos como la enfermedad de Parkinson [46]. Existen además otras características clínicas como disfonía, estridor laríngeo y distonía que presentaron algunos de nuestros pacientes pero que es más frecuente si se revisa la literatura, así lo demuestra una investigación donde la disartria ocurrió en la mayoría de los pacientes, alteración de los movimientos sacádicos (68%), estridor respiratorio (34%), antecollis (15%) y mioclonías (31%) [41].
Aún son pocos los estudios que han analizado el deterioro cognitivo en la AMS; quizás porque clásicamente se consideraba que la presencia de deterioro cognitivo en las fases iniciales del cuadro clínico era un criterio de exclusión para el diagnóstico de AMS. Sin embargo, estudios recientes demuestran que la presencia de tales trastornos incluso en fases iniciales de la enfermedad es perfectamente compatible con este diagnóstico (aunque no sea la forma de presentación típica) [8]. Estudios morfométricos por VBM (Voxel-based morphometry) demuestran que el deterioro cognitivo en estos pacientes se correlaciona tanto con la evolución de la enfermedad como con el grado de atrofia prefrontal. Este hallazgo coincide perfectamente con los estudios de neuroimagen funcional mediante SPECT, donde también se evidencia que el grado de deterioro neuropsicológico en pacientes con AMS-P se correlaciona con el descenso de perfusión cerebral a nivel prefrontal [47].
Dentro de los pacientes con AMS parecen existir diferencias en función del subtipo de AMS: los pacientes con AMS-P presentan empobrecimiento de la recuperación de información verbal, alteraciones visuoespaciales, baja fluencia verbal y síndrome disejecutivo, mientras que los pacientes con AMS-C tienen dificultad en el aprendizaje de nueva información y en mantener la atención sostenida [7,47]. Así, los pacientes con AMS-C puntúan peor en el test de Stroop y tardan más tiempo en realizar el trail-making test que los pacientes con AMS-P [48]. Con todo, de forma global se aprecia un deterioro cognitivo más extenso en pacientes con AMS-P que en pacientes con AMS-C [47].
Comparación de los trastornos neuropsicológicos en las α-sinucleinopatías
Como se ha mencionado en la introducción las α-sinucleinopatías presentan un importante grado de solapamiento clínico, por lo que en las últimas décadas se han estudiado pormenorizadamente las diferencias en los aspectos clínicos motores de estas enfermedades. Sin embargo se ha prestado escasa atención a las similitudes y diferencias que puedan existir en el plano cognitivo.
Existen estudios que comparan aspectos de la cognición entre la EP con demencia y la DCLewy. Unos estudios no encuentran diferencias globales en la cognición [48,49]. Otros encuentran que quizás pueda existir un déficit atencional y disfunción ejecutiva más marcado en la DLewy que en la DEP [50]
Respecto a su alteración neuropsicológica más característica, el trastorno visuoespacial, se ha visto que es similar en ambos procesos [51]. Incluso analizando la ejecución de test aislados que incluyen funciones ejecutivas y visuoespaciales las diferencias son escasas o inexistentes: test del reloj no se encontraron claras diferencias [52], test de la copia de pentágonos [53]. Brief Visual Retention Test [48], cancelación visual [48], discriminación visual y percepción de objetos [51], etc…
Las fluctuaciones en la atención se encuentran presentes tanto en la DCLewy como en la EP con demencia, si bien no se encuentran en la EP sin demencia [49].
El único estudio comparativo que existe hasta la fecha comparando valorando los aspectos cognitivos entre las tres sinucleinpatías [4] muestra que globalmente el mayor grado de deterioro cognitivo se encuentra en los pacientes con DCLewy. Los pacientes con DCLewy y AMS-P muestran un síndrome disejecutivo y afectación visuoespacial más marcada que lospacientes con EP, mientras que la afectación de la memoria está más severamente afectada en los pacientes con DCLewy que en la EP y la AMS [4]. A modo de resumen en la tabla 2 se recogen de modo comparativo las características clínicas de las α-sinucleinopatías, tomando como fuente los apartados previos de este trabajo, con especial incidencia en los trastornos neuropsicológicos.
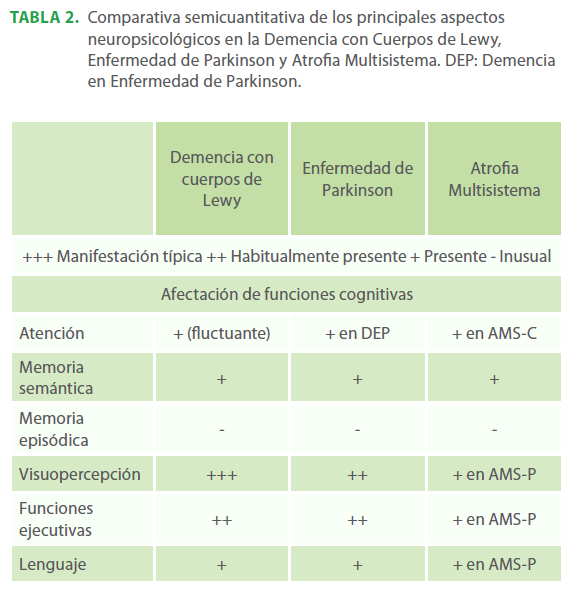
Tabla 2. Comparativa semicuantitativa de los principales aspectos neuropsicológicos en la Demencia con Cuerpos de Lewy, Enfermedad de Parkinson y Atrofia Multisistema. DEP: Demencia en Enfermedad de Parkinson.
A modo de conclusión podemos decir que los trastornos cognoscitivos forman parte importante del cuadro clínico de estas enfermedades. Estos trastornos pueden presentarse en grado variable y con patrones diferenciales en las distintas enfermedades neurodegenerativas por depósito de α-sinucleina. Aunque ya se han realizado algunos estudios de correlación clínico-patológica son necesarios más estudios neuropatológicos y de neuroimagen funcional para describir con precisión el sustrato neuropatológico de las alteraciones neuropsicológicas en las α-sinucleinopatías.
778
References
- Uéda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh. “Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease”. Proc. Natl. Acad. Sci. U.S.A. 1993;90 (23): 11282–6
- Tu PH, Galvin JE, Baba M, et al. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann Neurol 1998;44:415-422.
- Kao AW, Racine CA, Quitania LC, Kramer JH, Christine CW, Miller BL.Cognitive and neuropsychiatric profile of the synucleinopathies: Parkinson disease, dementia with Lewy bodies, and multiple system atrophy. Alzheimer Dis AssocDisord. 2009;23(4):365-70.
- Braak H, Rüb U, CansenSteur EN, Del Tredici K, De Vos RA. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 2005;64:1404-10
- Claassen DO, Josephs KA, Ahlskog JE, Silber MH, Tippmann- Peikert M, Boeve BF. REM sleep behavior disorder preceding other aspects of synucleinopathies by up to half a century. Neurology. 2010 Aug 10;75(6):494-9
- Balas M, Balash Y, Giladi N, Gurevich T. Cognition in multiple system atrophy: neuropsychological profile and interaction with mood. J Neural Transm. 2010;117(3):369-75
- Brown RG, Lacomblez L, Landwehrmeyer BG, Bak T, Uttner I, Dubois B, AgidY,Ludolph A, Bensimon G, Payan C, Leigh NP; NNIPPS Study Group. Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy. Brain. 2010;133(Pt 8):2382-93
- Aarsland D, Andersen K, Larsen JP, Lolk A, Kragh-Sorensen P. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol. 2003;60:387–392
- McKinlay A, Grace RC, Dalrymple-Alford JC, Roger D. Cognitive characteristics associated with mild cognitive impairment in Parkinson’s disease. Dement GeriatrCognDisord. 2009;28121-9
- Riedel O, Klotsche J, Spottke A, Deuschl G, Förstl H, Henn F, Heuser I, OertelW,Reichmann H, Riederer P, Trenkwalder C, Dodel R, Wittchen HU. Cognitive impairment in 873 patients with idiopathic Parkinson’s disease. Results from the German Study on Epidemiology of Parkinson’s Disease with Dementia (GEPAD). J Neurol. 2008;255(2):255-64
- Bronnick K, Emre M, Lane R, Tekin S, Aarsland D. Profile of cognitive impairment in dementia associated with Parkinson’s disease compared with Alzheimer’s disease. J NeurolNeurosurg Psychiatry. 2007;78, 1064-8
- Kulisevsky J, Pagonabarraga J. (2009) Cognitive impairment in Parkinson’s disease: tools for diagnosis and assessment. MovDisord. 24,1103-10
- Lee JE, Park HJ, Song SK, Sohn YH, Lee JD, Lee PH. Neuroanatomic basis of amnestic MCI differs in patients with and without Parkinson disease. Neurology 2010;75(22):2009-16
- Janvin CC, Larsen JP, Aarsland D, Hugdahl K. Subtypes of mild cognitive impairment in Parkinson’s disease: progression to dementia. MovDisord 2006; 21: 1343-9
- Mattila PM, Rinne JO, Helenius H, Dickson DW, Röyttä M. Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson’s disease. ActaNeuropathol. 2000;100(3):285-90
- Wolters EC, Braak H, Parkinson’s disease: premotor clinicophatological correlations. J NeurolTransmSuppl 2006; 70: 309-19
- Marinus J, Visser M, Verwey NA, Verhey FR, Middlekoop HA, Sttiggelbout AM, et al. Assessment of cognition in Parkinson’s disease. Neurology 2003; 61: 1222-8
- Ravina B, Marder K, Fernández HH, Friedman JH, McDonald W, Murphy D, et al. Diagnostic criteria for psychosis in Parkinson’s disease: report of an NINDS/NIMH Work Group. MovDisord 2007; 22: 1061-8
- Rodríguez-Ferreiro J; Cuetos F; Herrera E; Menéndez M; Ribacoba R. Cognitive impairment in Parkinson’s disease without dementia. Movement Disorders 2010;25(13):2136-41.
- Cuetos F, Menéndez M, Rodríguez-Ferreiro J. Semantic markers in the diagnosis of cognitive impairment in neurodegenerative dementias. Dement GeriatrCognDisord 2009;28:267-274
- Rodríguez-Ferreiro J, Menéndez M, Ribacoba R, Cuetos F. Action naming is impaired in Parkinson disease patients. Neuropsychologia 2009;47(14):3271-4
- Pillon B, Boller F, Levy R, Dubois B. Cognitive deficits and dementia in Parkinson’s disease. In Bolle F, Cappa S, eds. Handbook of neuropsychology. 2 ed. Amsterdam: Elsevier; 2001:311-71
- Kulisevsky J. Role of dopamine in learning and memory: implications for the treatment of cognitive dysfunction in patients with Parkinson’s disease. Drugs Aging 2000; 16: 365-79
- Kulisevsky J, Pagonabarraga J, Pascual-Sedano B, García-Sánchez C, Gironell A; Trapecio Group Study. Prevalence and correlates of neuropsychiatric symptoms in Parkinson’s disease without dementia.MovDisord. 2008 Oct 15;23(13):1889-96
- Verleden S, Vingerhoets G, Santens P. Heterogeneity of cognitive dysfunction in Parkinson’s disease: a cohort study. EurNeurol 2007; 58: 34-40
- McKeith I.G., Mosimann U.P. Dementia with Lewy bodies and Parkinson’s disease. Parkinsonism & Related Disorders 2004;10(Sup1):S15-S18
- Collerton D, Burn D, McKeith I, O’Brien J. Systematic Review and Meta-Analysis Show that Dementia with Lewy Bodies Is a Visual- Perceptual and Attentional-Executive Dementia. Dement GeriatrCognDisord 2003;16:229-237
- Kraybill ML, Larson EB, Tsuang DW, Teri L, McCormick WC, Bowen JD, Kukull WA, Leverenz JB, Cherrier MM. Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both Neurology, 2005 64:2069-2073
- Lambon Ralph et al. Semantic memory is impaired in both dementia with Lewy bodies and dementia of Alzheimer’s type: a comparative neuropsychological study and literature review J NeurolNeurosurg Psychiatry 2001;70:149-156
- Burton EJ, McKeith IG, Burn CJ, Williams ED, O’Brien JT. Cerebral atrophy in Parkinson’s disease, dementia with Lewy bodies and controls. Brain 2004; 127 (Pt 4): 791-800
- McShane R. Demencia en la enfermedad de Parkinson y demencia con cuerpos de Lewy. En: Jacoby R y Oppenheimer C coordinadores. Psiquiatría en el anciano. 1ª ed. Barcelona: Masson; 2005. p. 487-94.
- Shimomura T, Mori E, Yamashita H, Imamura T, Hirono N, Hashimoto M, Tanimukai S, Kazui H, HaniharaT.Cognitive Loss in Dementia With Lewy Bodies and Alzheimer Disease. Arch Neurol. 1998;55(12):1547-1552
- Simard, Martine, van Reekum, Robert, Cohen, Tammy. A Review of the Cognitive and Behavioral Symptoms in Dementia WithLewy Bodies J Neuropsychiatry ClinNeurosci 2000; 12: 425-450.
- Gómez-Tortosa E, Newell K, Irizarry MC, Albert M, Growdon JH, Hyman BT. Clinical and quantitative pathologic correlates of dementia with Lewy bodies. Neurology. 1999;53(6):1284-91
- Dejerine J, Thomas A. L’atrophieolivopontocerebelleuse. NouvIconogrSalpet 1900;13:330-370
- Shy GM, Drager GA. A neurological syndrome associated with orthostatic hypotension: a clinical-pathological study. Arch Neurol 1960;2:511-527.
- Graham JG, Oppenheimer DR. Orthostatic hypotension and nicotine sensitivity in a case of multiple system atrophy. J NeurolNeurosurg Psychiatry 1969;32:28-34.
- Robertson D, Shannon J, Jordan J, et al. Multiple system atrophy: new developments in pathophysiology and therapy. Parkinsonism RelatDisord 2001;7:257- 260
- Gilman S, Low PA, Quinn N, et al. Consensus statement on the diagnosis of multiple system atrophy. J NeurolSci 1999;163:94- 98.
- Wenning GK, Ben-Shlomo Y, Magalaes M, et al. Clinical features and natural history of multiple system atrophy: an analysis of 100 cases. Brain 1994;117:835-845
- Ballard CG, Aarsland D, McKeith I, O’Brien J, Gray A, Cormack F, Burn D, Cassidy T, Starfeldt R, Larsen JP, Brown R, Tovee M. Fluctuations in attention: PD dementia vs DLB with parkinsonism. Neurology 2002;59(11):1714-20
- Chrysostome V, Tison F, Yekhlef F, et al. Epidemiology of multiple system atrophy: a prevalence and pilot risk factor study in Aquitaine, France. Neuroepidemiol 2004;23:201-208.
- Sandroni P, Ahlskog J, Fealey R, et al. Autonomic involvement in extrapyramidal and cerebellar disorders. ClinAuton Res 1991;1:147-155.
- Riley D, Chelimsky T. Autonomic nervous system testing may not distinguish multiple system atrophy from Parkinson´s disease. J NeurolNeurosurg Psychiatry 2003;74:56-60.
- Kawai Y, Suenaga M, Takeda A, Ito M, Watanabe H, Tanaka F, Kato K, Fukatsu H, Naganawa S, Kato T, Ito K, Sobue G. Cognitive impairments in multiple system atrophy: MSA-C vs MSA-P. Neurology. 2008;70(16 Pt 2):1390-6
- Chang CC, Chang YY, Chang WN, Lee YC, Wang YL, Lui CC, Huang CW, Liu WL. Cognitive deficits in multiple system atrophy correlate with frontal atrophy and disease duration. Eur J Neurol. 2009;16(10):1144-50
- Noe, E., Marder, K., Bell, K. L., Jacobs, D. M., Manly, J. J. and Stern, Y. Comparison of dementia with Lewy bodies to Alzheimer’s disease and Parkinson’s disease with dementia. Movement Disorders 2004;19: 60–67
- Mondon K, Gochard A, Marqué A, Armand A, Beauchamp D, Prunier C, Jacobi D, de Toffol B, Autret A, Camus V, Hommet C. Visual recognition memory differentiates dementia with Lewy bodies and Parkinson’s disease dementia. J NeurolNeurosurg Psychiatry. 2007;78(7):738-41
- Mosimann MD. Visual perception in Parkinson disease dementia and dementia with Lewy bodies. Neurology 2004;63(11):2091-6
- Cahn-Weiner DA. Discrimination of Dementia With Lewy Bodies From Alzheimer Disease and Parkinson Disease Using the Clock Drawing Test. CognBehav Neurol. 2003;16(2):85-92
- Cormack F, Aarsland D, Ballard C, Tovée MJ. Pentagon drawing and neuropsychological performance in Dementia with Lewy Bodies, Alzheimer’s disease, Parkinson’s disease and Parkinson’s disease with dementia. Int J Geriatr Psychiatry. 2004;19(4):371-7.

