Keywords
Bothrombin; Inflammation; Biochemistry
Introduction
Snake venom serine proteases (SVSPs) are most important
and commonly used in the present era in the field of drug designing. This protease catalyze the cleavage of covalent bond
of peptides and act as important role in diverse biological
processes such as digestion, regulation of blood coagulation, inflammation, immunity etc. [1]. This protease are grouped into
six major clans and subsequently divided into families based on
their sequences (MER-OPS classification, https://
merops.sanger.ac.uk; [2]: exclusively clan SA, family S1. Among
all of them, SVSPs display specifically biological reactions in the blood coagulation cascade and platelet activation process.
Physiologically, SVSPs activate the prothrombin and
subsequently shorten the coagulation times. However, some
SVSPs also possess fibrinogen-clotting activity and act as
thrombin-like enzymes such as bothrombin, SVSP from
Bothropus jararaca.
Bothrombin was crystallized by the same lab [3], though no
three dimensional structure was made available so far.
Moreover, theoretical model of Bothrombin (1CXM), a serine
proteinase with fibrinogenolytic activity from the venom of
Bothrops jararaca was published by Serrano et al. [4].
Bothrombin form aggregates platelets in the presence of
fibrinogen and interact with glycoprotein 1b, which activates
blood cogulation factor VIII. Physiologically, it selective cleave
Arg-|-Xaa bond in fibrinogen, to form fibrin and release
fibrinopeptide A [5]. Thus, this enzyme encourages us to do
docking analysis with fibrinopeptide and get information about
its precise role in the blood coagulation process. This enzyme is
highly endemic and that is the reason many industries and
academic groups are looking for its inhibitor since the last
decade. The treatment usually falls into inhibitor such as diisopropyl
fluorophosphates but limited therapies due to serious
and life threatening side effects. At present, modern branches
such as biochemistry, molecular biology, genetics and
pharmacology have grown significantly in the ability to identify
specific biological targets [6]. In this continuation, computational
tools may add some fast and low-cost input to make data more
important to explore such targets in designing new drugs with
the aim to decrease fatality. Molecular docking and molecular
dynamics are two widespread used computational tools for
gaining insight into protein targets and their interactions and
dynamics at molecular level.
However, it has already been mention that bothrombin act as
thrombin like activity but drug interaction studies about
bothrombin inhibition assay are not found in the literature [7]. In
such case, fibrinopeptide with PDB code; 1DMK and 1YCP has
been used for docking study with bothrombin, which are 11 and
23 amino acid respectively [8].
In the present study, the 3-D structure of bothrombin has
been constructed using resulting FASTA sequences. Further, the
molecular model of bothrombin was constructed using
Modeller9v5 package for homology modeling using template
2AIP; protein C activator. The resulting model quality was
checked by using PROCHECK, PROSA, ERRAT, VERIFY 3D and
WHAT-IF and as a result on the overall scores, we choose the
final model. After final confirmation, protein-protein docking
was performed between the molecular models of bothromin
and fibrinopeptide (1DMK and 1YCP) by cluspro software. The
protein-protein docking analysis has also been done with these
peptides with protein C activator (2AIP) and thrombin (1BTH)
crystal molecule. At last, the binding energy in between all
complexes has been calculated by tinker software and analysis
the interpretation. The complete analysis of apparent inhibition
in addition to interaction of the models was carried out with
high binding affinity; make us inform that bothrombin act as
thrombin with more or less same catalytic activity. The resulting
result presented in this article will be useful to design molecules
for drug designing.
Methods
All the estimations were execute on a workstation Hi-end
server: Intel core 2 duo processor, 32 bits with 2 GB RAM with
video graphics card. Molecular modeling tasks were achieved
with Modeller9v5 (REF); protein-protein docking computations
were carried out with ClusPro server (REF). For homology
modeling of the Bothrombin, the crystal structure of Protein C
activator (PDB ID: 2AIP) from the venom of copperhead snake
Agkistrodon contortrix was used as a template and the docking
had been done with two fibrinopeptide; (1) PDB ID: 1DM4 chain
C (2) PDB ID: 1YCP chain F, with all default settings during
calculations.
Sequence alignments
Sequence of Bothrombin (gi No. AAB30013.1) was obtained
from National Centre for Biotechnology Information (NCBI). The
target sequence of bothrombin is used as a query sequence to
search for homologous protein structure(s) that could serve as
template(s) by running pblast [9] against PDB [10]. By using
blastp program template sequences of low similarity were
obtained. In order to enhance the query coverage we opted for
combination of multiple templates. Although the idea of
combining multiple templates sounds straightforward, its
implementation is fairly complex. The real challenge is not the
identification of a list of suitable template candidates, but an
optimal combination of these. This is because template search
methods ‘outperform’ the needs of comparative modeling in the
sense that they are able to locate so remotely related sequences
for which no reliable comparative model can be built. The
reason for this is that sequence relationships are often
established on short conserved segments, while a successful
comparative modeling exercise requires an overall correct
alignment for the entire modeled part of the protein. To find out
an appropriate template structure Protein C activator; 2AIP (gi
No. AAO85513.1) was selected, which has maximum identity.
The BLASTp alignment between selected templates was further refined using sequence alignments in the ClustalW2 (EMBL-EBI)
online software with default parameters [11].
Molecular model building
The search using the BLASTp alignment algorithm within the
PDB database showed various potential templates for molecular
modeling purposes. Only limited, crystallographic structures
were found to show high identity score with respect to
bothrombin sequence. Among all of them, PDB ID (2AIP) shows
maximum identity, this would be the reason; 2AIP was selected
as template for bothrombin for further studies. This sequence
was further refined using sequence alignment in the ClustalW
2.0.12 with default parameter. However, the 3D structure of
bothrombin was predicted by homology modeling on the basis
of the structure of by using the program Modeller 9v5. Modeller
9v5 is used to develop the structures for the proteins with
unknown structure by using comparative homology [12]: https://
salilab.org/modeller/tutorial/advanced.html]. A number of
models were generated by Modeller9v5 and were visualized
using Pymol. The modeling procedure begins with an alignment
of the sequence to be modeled (target) with relative known
three-dimensional structure (template). Many models were
generated and among them the one having lowest root mean
square deviation (RMSD) value when superimposed onto the
template 2AIP was chosen for further studies.
Validation of the homology model
After the construction of the model, its quality was assessed
considering both geometric and energetic aspects using
PROCHECK [13], ERRAT [14] and PROSA [15] for internal
consistency and reliability. The Ramachandran plot computed
with PROCHECK provided the residue position in particular
segment based on the dihedral angles. Finally, the best-quality
models were subjected to further calculations and molecular
modeling studies, binding site analysis and other calculations.
The structurally conserved regions (SCRs) between reference
proteins were then identified and superimposed, and then the
chosen models were subjected to energy minimization in order
to obtain a stable, low-energy conformation. We had generated
five models by Modeller, were further evaluated for quality by
calculating their energies. On evaluating the energies of these
models, Model 2 was found to have least energy value (DOPE
score) -14313.526367 (data not mention). This predicts the good
quality of Model 2 with relatively compact structure.
Protein-fibrinopeptide docking and analysis
In order to get insight into possible protein-fibrinopeptides
interactions, we have used six complexes formed between
thrombin (PDB code 1BTH), Protein C activator (PDB code 2AIP)
and energy minimized bothrombin with two fibrinopeptide (PDB
codes 1YCP and 1DM4). The docking was performed by
homology to the known complex conformation available in the
PDB by using PyMOL structural alignment tools. Following, the
initial complex coordinates were subject to energy minimization
in GROMACS.
To further compare each protein-peptide complex, the
electrostatic potential complementarity between the proteininhibitor
complexes were also checked using the pdb2pqr web
server [16] and APBS software [17]. The electrostatic potential
surface were generated and compared to the experimentally
solved thrombin and protein C activator.
Binding affinity
To further compare the studied complexes, we assessed the
energetics of each resulting protein-peptide complex. The
TINKER Molecular Modeling Package [18] was used to energy
minimize each structure (monomers subunits and complexes)
and to compute the binding energy components. By using the
same model as REF, we state that the binding energy may be
estimated by:
ΔGbind=Gcomplex-Gprotein-Gpeptide
Where the thermodynamic potential G may be approximate
to the bonding, angle, van der Waals and Coulombic terms:
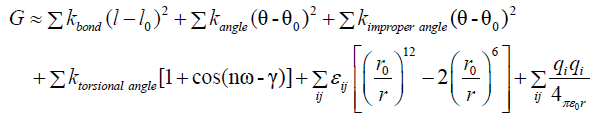
Where each constant are taken for the selected force field in
TINKER Molecular Modeling Package. For this study we used the
AMBER force field [19].
Virtual alanine scanning
In order to get a more detailed picture of the most important
residues for establishing the protein-protein interaction
interface, we used computational alanine scanning. Robetta
Alanine Scanning [20,21] was employed for such a task. In this
protocol, each amino acid residue is replaced by alanine
residues. The difference in the binding energy (as calculated by
Robetta Alanine Scanning algorithm) is then associated to the
amino acid importance. Amino acid residues which contribute
with more than 2 kcal/mol for the protein-protein complex
binding energy is referred to as hot spot.
Results
Sequence alignments
To attain an appropriate template structure for assembling
the target pattern, the sequence of bothrombin was retrieved
from NCBI (National Center for Biotechnology Information). To
get a high-quality template for homology modeling of
bothrombin, the amino acid sequence was aligned against the
Protein Data Bank [22]. This alignment was executed by means
of the BLAST algorithm. Subsequently, further refinements of
these sequences alignment in the ClustalW 2.0.12 with default restrictions were performed. Multiple sequence alignment
informs that the 2AIP and bothrombin are 71% similar in nature and have major difference in the amino acid sequence on the
turn and bend region as shown in Figure 1, performed by
ClustalW 2.0.12. This information also support that 2AIP was
selected as good template for modeling of Bothrombin. The
figures informs that the residues involved in binidng of various
feedback inhibitors in template were also been conserverd in
the Bothrombin (data not shown).
Figure 1: Sequence alignment of bothrombin and protein c
activator (2AIP) ClustalW 2.0.12. Highly conserved residues
are represented in star.
Molecular model building
The exploration by the BLASTp alignment algorithm within the
PDB database illustrates impending templates for molecular
modeling objects. Selection of appropriate template was chosen
based sequence similarity, residue compactness, and crystal
resolution [23]. Many structures were found to show high
identity score with respect to bothrombin but among them,
crystal structure of native protein C activator (2AIP) from the
venom of copperhead snake Agkistrodon contortrix (PDB code:
2AIP, resolution 1.65 Å, R-value 0.17086 (obs), and R-Free
0.19692) [16] was selected as template structure. The 3D
structures bothrombin was predicted by homology modeling on
the basis of the structures of 2AIP by using the program
Modeller 9v5. This software is an automated attempt to
comparative modeling, begins with an alignment of the
sequence to be modeled as target with relative known threedimensional
structures act as templates. Many models were
generated and among them the one having lowest root mean
square deviation (RMSD) value was selected as template for
further analysis [24]. At last, the chosen models were subjected
to energy minimization to get the most stable conformation for
our study.
Validation of the homology model
The quality of the constructed model was evaluated by both
geometric and energetic aspects using PROCHECK, ERRAT and
PROSA for internal uniformity and consistency. The first
validation was carried out using Ramachandran plot analysis
computed with PROCHECK. PROCHECK has been used to provide residue position based on the dihedral angles in the form of
Ramachandran plot by checking residue-by-residue
stereochemical quality of the protein structure [25]. The analysis
showed that 2AIP, energy minimized Bothrombin and theoretical model of Bothrombin (1CXM) in the most favorable region were
83.3%, 78.2%, and 74.1% and in the additional allowed region
15.6%, 17.6%, and 22.8%, respectively (Table 1 and Figure 2A and 2B).
| Structure |
Average package qualitya |
Rotamer normalityb |
Backbone conformation |
Bond lengthc |
Angled |
| 2AIP |
0.086 |
0.539 |
-5.278 |
0.478 |
0.718 |
| Bothrombin |
-0.54 |
-2.555 |
-6.319 |
0.67 |
1.233 |
| 1CXM |
-0.82 |
-1.975 |
-10.199 |
1.234 |
1.419 |
| 1BTH_H |
-1.418 |
-1.637 |
-5.636 |
0.475 |
0.753 |
(a) The average quality of 200 highly refined X-ray structures was -0.5 ± 0.4
(b) The behaviour of the distribution is much that a Z-score below -2 (2 standard deviations way from the average) is poor and a Z-score of less than -3 is of concern; positive is better than average
(c)RMSD Z-score should be close to 1.0
(d)RMSD Z-score, more common values are around 1.55
Table 1: What if stereochemical quality evaluation.
Figures 2A and B: Ramachandran plot of developed.
Ramachandran plot by checking residue-by-residue
stereochemical quality of the protein structure. The analysis
showed that 2AIP, energy minimized Bothrombin and most
favorable region were 83.3%, 78.2%, and 74.1% and in the
additional allowed region 15.6%, 17.6%, and 22.8%,
respectively.
PROCHECK has been used to provide the best-quality models,
which were selected for further molecular modeling studies,
binding site analysis and some other necessary information. The
structurally conserved regions (SCRs) between suggested
proteins were then recognized and superimposed as a result the
bothrombin sequence was then aligned to the SCRs using the
alignment module. The superimposition of 2AIP and bothrombin
has shown here in Figures 3A and 3B where final modeled
structure of bothrombin is composed of 13 beta-strands
arranged into two domains, similarly to 2AIP structure, and two
long alpha-helixes and short one near the catalytic site (Figure
4). The catalytic region lies on the surface between the two
beta-strands motifs. The catalytic triad (His 57, Asp102, and
Ser195) formed in the 2AIP have similar orientation as compare
to the catalytic residues His-41, Asp-86 and Ser-178 of
bothrombin are highlighted in Figures 5A and 5B. The
superimposition of bothrombin did not show major structural conformational changes in comparison to the template model;
2AIP, which in turn is consistent with the relatively low RMSDF
values. The overall low RMSD values reflect the high structural
conservation of this complex through evolution, making it a
good system for homology modeling (Table 2).
Figures 3A and B: The modeller structure of the
superimposition of 2AIP and bothrombin.
Figure 4: Secondary structure of template 2AIP and
bothrombin.
Figures 5: (a) The ERRAT score for the bothrombin model is 80.09, the backbone conformation PROSA-web Z-scores of all protein
chains in PDB determined by X ray crystallography (light blue) and NMR spectroscopy (dark blue) with respect to their length (b)
ERRAT score for bothrombin.
| PROCHECK |
Z score |
ERRAT Score |
| Ramachandran plot quality (%) |
Goodness factor |
| |
Most favored |
Additional allowed |
Generously allowed |
Dis-allowed |
Overall |
| 2AIP |
83.3 |
15.6 |
1 |
0 |
0 |
-6.57 |
83.019 |
| Bothrombin |
78.2 |
17.6 |
3.1 |
1 |
-0.4 |
-6.7 |
80.097 |
| 1CXM |
74.1 |
22.8 |
2.1 |
1 |
-0.6 |
-7.11 |
55.051 |
Table 2: PROCHECK and ERRAT.
By applying the structural superposition and RMSD
evaluations, our model appears very similar to the theoretical
model, 1CXM but with good quality assessment analysis. The
total quality G-factor -0.4 was calculated (average value are in
between 0 and -0.5), which informs that the model was good in
structure. However, G-factor value of 1CXM was -0.6; consider
being not a good model for further analysis.
However, PROCHECK analysis showed no bad scores for side
chain and main chain parameters for both bothrombin and
1CMX, but has better quality for bothormbin. In addition for
non-bonded atomic interactions, ERRAT score is consider as
overall quality factor and higher value (>50) consider as better
quality. In the current case, the ERRAT score for bothrombin was
80.097, which fit well within the high quality model as shown in Figure 5A. However, the ERRAT score of 1CXM was 55.051,
which in comparison to support that the bothrombin model is
more perfect for further analysis as shown in Table 1.
In the analysis, PROSA Z-score was used to judge the
interaction energy of each residue with the remainder of a
protein was computed whether it fulfills certain energy criteria
or not. Evaluation of the energy minimized model of bothrombin
with PROSA web revealed that Z-score value was -6.7 as shown
in Table 1 and Figure 5B. This indicates no significant deviation
from typical native structure as the template when compared with Z-score of -6.57 for 2AIP template. However, the Z-score of
1CXM was -7.11, informs that modeled bothrombin have better
homology with 2AIP in comparison to 1CXM. Thus the
bothrombin structure was found to be reasonable and reliable
conformation for further protein docking studies.
Protein-protein docking
I need these PDBs of the complexes that will be analyzed. In
order to elucidate the inhibition mechanism on bothrombin, two
fibrinopeptide (1dm4 and 1ycp) were used as a ligand for
docking study to evaluate the productive ability for use in the
computational structure-based protein-protein studies. As
reported, bothrombin behave like thrombin but due to
limitation of its structure, we performed modeling of
bothrombin by using fasta sequence as reported. Fasta
sequence of bothrombin has similarity (more or less similar
RMSD) with 1BTH and 2AIP as mention in the (Figure 6A),
encourage us to select thrombin as a template. But, among both
of them 2AIP have high resolution (1.65 Å) than 1BTH (2.30 Å),
which give it more preference. The electrostatic potential
surface as shown in Figure 6B, inform that active site of
bothrombin have negative charges surrounded by positive
charges as similar in case of 1BTH rather than 2AIP. This
information encourage us that botrombin would behave like thrombin and have play important role in degradation of
fibrinogen. However, as shown in Figure 6C, the active site
pocket of bothrombin is more similar to 2AIP than 1BTH, informs
bothrombin have similar selection of substrate as 2AIP. This all
information motivates to do docking of bothrombin, 2AIP and
1BTH with fibrinopeptide (1YCP and 1DM4). To perform the
protein-protein docking between all complexes, ClusPro server
was used in its default parameters, and potential attractions
between the interface residues, as assessed in the
experimentally solved complex were employed. The ten best
representatives of different clusters were compared, achieving on average 1 Å RMSD and up to 5 Å RMSD (Choose a threshold
less than 5A (2 or 3A I think) so that it has only 4 structures for
each case, it would be very expensive experimentally to test 10
cases for each protein-ligand). Further, all six complexes
(2AIP_1DM4, 2AIP_1YCP, 1BTH_1DM4, 1BTH_1YCP,
Bothrombin_1DM4 and Bothrombin_1YCP) are further use for
binding energy calculations as shown in Figure 7A-7C. This
informs that 2AIP_1DM4 and 2AIP_1YCP have less ΔG value in
comparison to rest of the 1DM4 and 1YCP complexes,
respectively (Table 3).
Figure 6: Docking of bothrombin, 2AIP and 1BTH with
fibrinopeptide (1YCP and 1DM4). To perform the proteinprotein
docking between all complexes, ClusPro server was
used in its default parameters, and potential attractions
between the interface residues.
Figure 7: Six complexes (2AIP_1DM4, 2AIP_1YCP,
1BTH_1DM4, 1BTH_1YCP, Bothrombin_1DM4 and
Bothrombin_1YCP) are further use for binding energy
calculation.
| |
Bond Stretching |
Angle Bending |
Improper Torsion |
Torsional Angle |
Van der Waals |
Charge-Charge |
Implicit Solvation |
Total Potential energy |
Intermolecular Energy |
| Bothrombin |
322.2461 |
708.8676 |
34.6025 |
2295.1609 |
-838.9149 |
-7299.3628 |
-1375.505 |
-6152.9056 |
|
| 2AIP |
166.7472 |
499.3745 |
16.3666 |
2026.5276 |
-1246.6104 |
-8740.7519 |
-2943.3916 |
-10221.7381 |
-1617.7321 |
| 1BTH |
157.6084 |
516.6546 |
17.5501 |
2181.1625 |
-1480.1138 |
-6998.2166 |
-4901.7227 |
-10507.0775 |
-454.3891 |
| 1DM4_C |
7.0249 |
11.7386 |
0.3714 |
54.9343 |
-15.9343 |
-421.0023 |
-270.2253 |
-633.0875 |
-32.0757 |
| 1YCP_F |
5.365 |
11.0937 |
0.1313 |
35.9067 |
1.5188 |
-289.9766 |
-213.2131 |
-449.1744 |
-31.4309 |
| 1BTH_1DM4 |
189.0033 |
650.0719 |
42.9134 |
2439.375 |
-1247.2866 |
-10403.9477 |
-2279.6641 |
-10609.5347 |
-663.0243 |
| 1BTH_1YCP |
1382.9255 |
684.9666 |
47.982 |
2440.9696 |
-641.9088 |
-9735.8124 |
-2738.7649 |
-8559.6423 |
-625.1846 |
| 2AIP_1DM4 |
190.1015 |
544.5895 |
32.58 |
2206.5655 |
-1017.0955 |
-11395.7499 |
-814.3604 |
-10253.3693 |
-1665.63 |
| 2AIP_1YCP |
191.1646 |
537.8985 |
32.5679 |
2189.6982 |
-980.847 |
-10878.0231 |
-1143.7864 |
-10051.3273 |
-1606.4641 |
| both_1DM4 |
131.493 |
587.26 |
42.0334 |
2307.9445 |
-1259.988 |
-7993.655 |
-1279.9834 |
-7464.8964 |
-135.4541 |
| both_1YCP |
133.4075 |
595.3572 |
38.0078 |
2281.1657 |
-1216.1594 |
-7790.8477 |
-1351.1726 |
-7310.2415 |
-97.3496 |
Table 3: Binding energy calculation of different complexes of docking studies.
Discussion
The BLASTp result revel that bothrombin sequence has high
sequence and structure similarity with 2AIP, which depicts that,
the conserved amino acids intricate in the binding of active site
catalytic triad (His 57, Asp102, and Ser195). The catalytic site
region as shown as bobble are also shown in the Figure 6,
informed that bothrombin share the same pocket as 2AIP. The
bothrombin also have similar electrostatic potential with 1BTH
(thrombin) as shown in Figure 6, inform that it would behave as
thrombin like protein and act as role in degradation of
fibrinogen. All these proteins viz, bothrombin, 2AIP, 1BTH were
share lowest RMSD value, which finally select bothrombin as
model for this study. Bothrombin was modeled by Modeller 9v5
and the final selected bothrombin model, the energy
minimization was calculated by Tinker software to elucidate the
most stable conformation for study. The quality of the model
was appraised by PROSA, PROCHECK, ERRAT, and WHAT IF for
their consistency and stability as also reported. The PROSA was
used to calculate the interaction energy per residue and
evaluate whether it fulfills certain energy criteria or not. The
PROSA Z-Score is their output result, which analyzed. ERRAT on
the other hand consider as overall quality factor and its higher
score mean better quality.
As reported, the bothrombin ERRAT score is more than
80.097, which inform that the consider model is one of the best one. The model has high similarity with theoretical model 1CXM
after calculated by PROCHECK analysis as shown in Table 1 and Figure 2A and 2B. All information suggests that the bothrombin
behave as thrombin and to do further docking with
fibrinopeptide were analyzed as reported. The results were
shown in Figure 7, which informed that 2AIP, 1BTH and
bothrombin are able to degrade the fibrinopeptide. Similar
analysis with three dimensional interactions also reviewed.
To elucidate the interaction as wet lab would be a very
expensive process, in such context; this study wills
empowerment to biochemist and pharmacologist to use
bothrombin as one of the substitute of Thrombin in case of
fibrinolytic degradation. The generated bothrombin model is
anticipated to be useful for structure based drug designing, in
addition.
References
- Neurath H (1984) Evolution of proteolytic enzymes. Science 224: 350-357.
- Rawlings ND, Tolle DP, Barrett AJ (2004) MEROPS: the peptidase database. Nucleic Acids Res 32: D160-164.
- Watanabe L, Vieira DF, Bortoleto RK, Arni RK (2002) Crystallization of bothrombin, a fibrinogen-converting serine protease isolated from the venom of Bothrops jararaca. Acta Crystallogr D Biol Crystallogr 58: 1036-1038.
- Serrano SM, Maroun RC (2005) Snake venom serine proteinases: sequence homology vs. substrate specificity, a paradox to be solved. Toxicon 45: 1115-1132.
- Nishida S, Fujimura Y, Miura S, Yoshida E, Sugimoto M, et al. (1994) Purification and Characterization of Bothrombin, a Fibrinogen-Clotting Serine Protease from the Venom of Bothrops jararaca. Biochemistry 33: 1843-1849.
- Vivas-Ruiz DE, Sandoval GA, Mendoza J (2013) Coagulant thrombin-like enzyme (barnettobin) from Bothrops barnetti venom: Molecular sequence analysis of its cDNA and biochemical properties. Biochimie 95: 1476-1486.
- Watanabe L, Vieira DF, Bortoleto RK, Arni RK (2002) Crystallization of bothrombin, a fibrinogen-converting serine protease isolated from the venom of Bothrops jararaca. Acta Crystallogr D Biol Crystallogr 58: 1036-1038.
- Henriques ES, Fonseca N, Ramos MJ (2004) On the modeling of snake venom serine proteinase interactions with benzamidine-based thrombin inhibitors. Protein Sci 13: 2355-2369.
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402.
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, et al. (2000) Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47: 351-354.
- Thompson JD, Higgins DG, Gibson TJ (1994) Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673-4680.
- Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M (1995) Evaluation of comparative protein modeling by modeller. Proteins 23: 318-326.
- Laskowski RA, MacArthur MW, Moss DS, Thornton TM (1993) PROCHECK: A program to check the stereochemical quality of protein structures. J Applied Cryst 26: 283-291.
- Colovos C, Yeates TO (1993) Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci 2: 1511-1519.
- Sippl MJ (1993) Recognition of errors in three-dimensional structures of proteins. Proteins 17: 355-362.
- Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, et al. (2007) PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations, Nucleic Acids Res 35: 522-525.
- Unni S, Huang Y, Hanson RM, Tobias M, Krishnan S, et al. (2011) Web servers and services for electrostatics calculations with APBS and PDB2PQR. J Comput Chem 32: 1488-1491.
- Guex N, Peitsch MC (1997) Swiss Model and the Swiss- PdbViewer: an environment for comparative protein modeling. Electrophoresis 18: 2714–2723.
- Nelson J (2017) Tinker - Software Tools for Molecular Design.
- Kortemme T, Kim DE, Baker D (2004) Computational alanine scanning of protein-protein interfaces. Sci STKE 2004: 2.
- Kortemme T, Baker D (2002) A simple physical model for binding energy hot spots in protein-protein complexes. Proc Natl Acad Sci U S A 99: 14116-14121.
- Colovos C, Yeates TO (1993) Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci 2: 1511-1519.
- Castro HC (2001) Structural features of a snake venom thrombin-like enzyme: thrombin and trypsin on a single catalytic platform. Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology 47: 183-195.
- Harold AS (2004) The thrombin–fibrinogen interaction. Biophys Chem 112: 117-130.
- Thierry R, Enrico DC (2002) Three-dimensional Modeling of Thrombin-Fibrinogen Interaction. Journal of Biological Chemistry 277: 18875-18880.