Keywords
?rterial hypertension; Heart failure; Extracellular matrix
Introduction
Arterial hypertension (AH) is a leading cause for large number of heart failure cases [1]. It is well known that when investigating cellular and molecular biology of heart failure, scientific research was long time focused on cardiomyocyte dysfunction [2].
Extracellular matrix (ECM) consists of collagen fibrils, elastin fibers, fibroblasts [3]. Macrophages, macromolecules such as glycoproteins, glycosaminoglycans together with other molecules such as growth factors, cytokines and extracellular proteases also take part in this structure’s props. The collagen matrix in the myocardium may be conceptualized as scaffolding in which collapse of the individual components of the scaffold may not necessarily result in an absolute loss of collagen content but rather in a loss of structural integrity [4].
Traditionally cardiac ECM is thought to be a relatively inactive structure, which is playing a role of cardiac myocytes and vessels’ scaffold. Recent data provides evidence that cardiac ECM is a dynamic and metabolically active structure. Changes in cardiac extracellular matrix are suspected of contributing to the genesis and progression of heart failure [5].
Myocardial remodeling is a main mechanism in pathophysiology of chronic heart failure (CHF). This process includes two main stages at cellular level (1) cardiomyocyte hypertrophy, dysfunction, and necrosis and (2) increased deposition and damage of ECM, frequently described by the term “myocardial fibrosis“. Pathological collagen turnover and increased synthesis and deposition of collagen type I and III in myocardium takes main role in myocardial fibrosis. This process is regularized by matrix metalloprotease MMP-1,2,3,8,13 and tissue inhibitor of matrix metalloprotease TIMP-1 [6].
Elastin is a connective tissue protein with highly elasticity. It is a major ECM component and allows many tissues in the body to resume their shape after stretching or contracting. Elastin is involved in cardiac ECM structure and function [7]. Elastin turnover is regularized mainly by MMP-12 ? TIMP-3.
To this moment, there are insufficient data of elastin turnover in CHF. Investigation of elastin turnover should help for better understanding the role of ECM in pathogenesis of CHF. Abnormal changes in cardiomyocytes structure might not be the only one pathophysiological mechanism involved in development and progression of heart failure [8]. That is why the focus of our study is myocardial ECM. This might help for better understanding and future therapies in CHF.
According to left ventricular ejection fraction value HF can be classified as
• HF with preserved EF (HFpEF); normal LVEF [typically considered as ≥ 50%]
• HF with reduced EF (HFrEF); reduced LVEF [typically considered as ≤ 40%]
• HF with mid-range ejection fraction EF (HFmrEF); [patients with an LVEF in the range of 40-49% represent a ‘grey area’]
HFmrEF is a syndrome, defined by: (1) LVEF%-40-49%, (2) Symptoms and/or signs of heart failure, (3a) Elevated levels of natriuretic peptides, (3b) At least one additional criterion: relevant structural heart disease (LVH/left atrial enlargement) or diastolic dysfunction- 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure [9].
The aim of our study was to (1) Measure levels of matrix metalloprotease-12 (MMP-12) and tissue inhibitor of matrix metalloprotease -12 (TIMP-3) in patients with AH and HFmrEF and (2) to compare their levels with healthy controls.
“Matrix metalloproteases (MMPs) comprise a family of enzymes that cleave protein substrates based on a conserved mechanism involving activation of an active site-bound water molecule by a Zn2+ ion. MMPs family are involved in the breakdown of extracellular matrix in normal physiological processes, such as embryonic development, reproduction, and tissue remodeling, as well as in disease processes, such as arthritis and metastasis.
They play central roles in morphogenesis, wound healing, tissue repair and remodelling in response to injury, e.g. after myocardial infarction, and in progression of diseases such as atheroma, arthritis, cancer and chronic tissue ulcers. They are multi-domain proteins and their activities are regulated by tissue inhibitors of metalloproteinases (TIMPs)” [10].
“Matrix metalloproteinase-12 (MMP-12) also known as macrophage metalloelastase (MME) or macrophage elastase (ME) is an enzyme that in humans is encoded by the MMP12 gene. As the name reveals, a major substrate for MMP-12 is elastin, but MMP-12 is capable of degrading other ECM constituents (but not gelatin) and many nonmatrix proteins in vitro. Most MMP's are secreted as inactive proproteins. The enzyme degrades soluble and insoluble elastin” [11,12]. MMP12 may play a role in aneurysm formation and Stanford-A acute aortic dissection [13,14] and studies in mice and humans suggest a role in the development of emphysema [15]. MMP-12 plays role in tissue remodeling in Fas-Induced Lung Fibrosis Gustavo [16], and in fibrotic diseases, including systemic sclerosis (SSc) and correlates with vasculopathy and fibrosis potentiating skin and heart fibrosis in these patients [17]. Matrix metalloproteinase 12-deficiency augments extracellular matrix degrading metalloproteinases and attenuates IL-13–dependent fibrosis in the liver and lung of mice in response to S. mansoni eggs, confirmed by both DNA microarray and real-time PCR analysis. In the study, they reveal that MMP12 acts as a potent inducer of inflammation and fibrosis after infection with the helminth parasite S. mansoni [18]. Moreover it is observed interactions between antihypertensive drugs and MMP-9 and MMP-12 for coronary heart disease and composite CVD [19]. The data suggest that MMP9 R668Q (rs2274756), MMP9 R279Q (rs17576), MMP12 N122S (rs652438) genes and MMP12 N122S locus may provide useful clinical information with respect to treatment decisions.
Tissue inhibitor of metalloproteinases (TIMPs) inhibits matrix metalloproteinases (MMPs) that degrade the matrix structural proteins. MMP-12 is inhibited by TIMP-3. In response to a hypertensive stimulus, the balance between MMPs and TIMPs is altered. There is insufficient data in literature for MMP-12 and TIMP-3 turnover in patients with essential hypertension and heart failure with mid-range ejection fraction till this moment.
As per the examine polymorphisms in the promoter regions of MMP-2, MMP-3, MMP-9 and MMP-12 genes as determinants of aneurysmal coronary artery disease and found that the MMP- 3 5A allele is associated with the occurrence of coronary aneurysms. Their results suggest that an increased proteolysis in the arterial wall may act as a susceptibility factor for the development of coronary aneurysms in patients with coronary atherosclerosis [20].
Hypothesized that after an aortic stenosis MMP-2 released angiogenic factors during compensatory hypertrophy and MMP-9/ TIMP-3 released anti-angiogenic factors causing decompensatory heart failure [21]. To verify this hypothesis, they study wild type (WT) mice 3 and 8 weeks after aortic stenosis, created by banding the ascending aorta in WT and MMP-9-/- (MMP-9KO) mice.
Cardiac function (echo, PV loops) was decreased at 8 weeks after stenosis. The levels of MMP-2 (western blot) increased at 3 weeks and returned to control level at 8 weeks, MMP-9 increased only at 8 weeks. TIMP-2 and -4 decreased at 3 and even more at 8 weeks. The angiogenic VEGF increased at 3 weeks and decreased at 8 weeks, the anti-angiogenic endostatin and angiostatin increased only at 8 weeks. CD-31 positive endothelial cells were more intensely labelled at 3 weeks than in sham operated or in 8 weeks banded mice. Vascularization, as estimated by x-ray angiography, was increased at 3 weeks and decreased at 8 weeks postbanding. Although the vast majority of studies were performed on control WT mice only, interestingly, MMP9-KO mice seemed to have increased vascular density 8 weeks after banding. These results suggested that there was increase in MMP-2, decrease in TIMP-2 and -4, increase in angiogenic factors and vascularization in compensatory hearts. However, in decompensatory hearts there was increase in MMP-9, TIMP-3, endostatin, angiostatin and vascular rarefaction.
In Yasmin et al. [22] hypothesized that elastase activity would be related to arterial stiffness and tested this using isolated systolic hypertension (ISH) as a model of stiffening and separately in a large cohort of healthy individuals. A total of 116 subjects with ISH and 114 matched controls, as well as 447 individuals free from cardiovascular disease were studied. Aortic and brachial pulse wave velocity (PWV) and augmentation index were determined. Blood pressure, lipids, C-reactive protein, MMP- 9, MMP-2, serum elastase activity (SEA), and tissue-specific inhibitor 2 of metalloproteinases were measured. Aortic and brachial PWV, MMP-9, MMP-2, and SEA levels were increased in ISH subjects compared with controls (P=0.001). MMP-9 levels correlated linearly and significantly with aortic (r=0.45; P=0.001) and brachial PWV (r=0.22; P=0.002), even after adjustments for confounding variables. In the younger, healthy subjects, MMP-9 and SEA were also independently associated with aortic PWV. Authors conclude that aortic stiffness is related to MMP-9 levels and SEA, not only in ISH, but also in younger, apparently healthy individuals. This suggests that elastases including MMP-9 may be involved in the process of arterial stiffening and development of ISH. The relationship between arterial stiffness and elastase activity was examined in isolated systolic hypertension (ISH), and separately in a large cohort of healthy individuals. Aortic stiffness is related to MMP-9, not only in ISH, but also in healthy individuals, suggesting elastases may be involved in the process of arterial stiffening and the development of ISH.
The current advances of collagen quantification methods are who present a unique quantitative algorithm to quantify not only distribution but also abundance of gap junctions from cardiac tissue sections [23]. Their main innovation involved development of a quantitative blur algorithm that used N-Cad as an internal reference to quantify both distribution and abundance of Cx43 from Cx43/N-Cad double immuno-stained cardiac tissue sections. In addition, they have developed a computer-based collagen quantification algorithm to collectively quantify interstitial collagen, excluding unstained empty areas on Masson's Trichrome stained histological slides.
Authors Yan et al. [24] study potential contribution to various cardiac conditions including arrhythmia and report for the first time that aged rabbit LA exhibited significantly enhanced c-Jun N-terminal kinase (JNK) signaling that was associated with significantly reduced gap junction connexin43 (Cx43) and increased atrial arrhythmogenicity. The direct aetiological role of JNK activation was supported by their observations that the treatment of young rabbits with the JNK activator anisomycin led to increases of activated JNK, reductions of Cx43, and increases of pacing-induced AT/AF similar to those found in aged LA. Using cultured HL-1 atrial monolayers, they further demonstrated the pivotal role of JNK activation on Cx43 suppression and arrhythmogenicity evident from the restored amount of Cx43 and prevention of slowed CV and arrhythmogenicity by SP600125 specific JNK inhibition. Their results strongly suggest an important role of JNK activation in the development of AF.
Authors Westmann et al. [25] investigated mining novel constitutive promoter elements in soil metagenomic libraries in Escherichia coli. They experimentally mined novel constitutive promoter sequences in metagenomic libraries by combining a bidirectional reporter vector, high-throughput fluorescence assays and predictive computational methods. Through the expression profiling of fluorescent clones from two independent soil sample libraries, we have analyzed the regulatory dynamics of 260 clones with candidate promoters as a set of active metagenomic promoters in the host Escherichia coli. Through an in-depth analysis of selected clones, they were able to further explore the architecture of metagenomic fragments and to report the presence of multiple promoters per fragment with a dominant promoter driving the expression profile. These approaches resulted in the identification of 33 novel active promoters from metagenomic DNA originated from very diverse phylogenetic groups. The in silico and in vivo analysis of these individual promoters allowed the generation of a constitutive promoter consensus for exogenous sequences recognizable by E. coli in metagenomic studies. The results authors present demonstrate the potential of functional metagenomics for exploring environmental bacterial communities as a source of novel regulatory genetic parts to expand the toolbox for microbial engineering.
Methods
Ethics
All procedures were in accordance with the ethical standards of the responsible committee on human experimentation (institutional or regional) and with the Helsinki Declaration of 1975, as revised in 2000 (available at https://www.wma.net/e/policy/17-c_e.html). Approval of local Ethics Committee was obtained and informed consent from adult research participants was obtained too.
56 patients with AH and HfmrEF were examined, mean age 65.62 ± 9.69 (Group 1); and healthy subjects for controls (n=12), mean age 56.4 ± 5.53 (Group 2). 41 of patients were with target organ damage and 15 were without (Table 1).
• Serum total cholesterol and triglyceride concentrations were measured by enzyme assay (Boehringer Mannheim, Mannheim, Germany).
• Arterial blood pressure was measured using a standard anearoid sphygmomanometer, to the nearest 2 mm Hg, in the dominant arm after at least 10-min rest in supine position.
• Enzyme-linked immunosorbent assay (ELISA) Kits was used for measuring MMP-12 and TIMP-3 levels.
- (NV-DDXK-E-MMP12-1) Human MMP-12 ELISA kit, 96-well plate- Novus Biologicals
- (NV-KA0420) TIMP-3 human ELISA kit, 96-well plate- Novus Biologicals
• Echocardiographic methods. Echocardiography was performed with GE (Vivid) with 4-MHz transducer. All measurements were obtained according to European Society of Cardiology (ESC) criteria (Table 2).
?able 1 Clinical data of patients.
| Clinical data |
Patients |
Controls |
| Age |
64.07 ± 10.58 |
56.4 ± 5.53 |
| Gender (M/F) |
36/20 |
8/4 |
| Mean duration AH |
8.6 ± 5.9 |
N/A |
| Mean duration HF |
5.6 ± 2.9 |
N/A |
| SBP (mmHg) |
145.23 ± 17.23 |
118.26 ± 13.74 |
| DBP (mmHg) |
86.73 ± 11.03 |
76.4 ± 8.4 |
| BMI |
28.44 ± 5.48 |
22.11 ± 3.27 |
| Total cholesterol (mmol/l) |
4.89 ± 1.06 |
3.99 ± 0.65 |
| HDL (mmol/l) |
1.01 ± 0.25 |
0.96 ± 0.20 |
| LDL (mmol/l) |
3.14 ± 1.12 |
2.43 ± 0.64 |
| Triglycerides (mmol/l) |
1.49 ± 1.32 |
1.31 ± 0.61 |
| Blood glycose |
6.05 ± 0.68 |
N/A |
| Creatinine |
100.32 ± 3.28 |
N/A |
| Hypertensive heart damage |
(n=21) |
|
| Hypertensive brain damage |
(n=6) |
|
| Hypertensive kidney damage |
(n=10) |
|
| Hypertensive eye damage |
(n=4) |
|
| Smokers |
32 |
|
| Count |
56 |
12 |
Table 2 Echocardiographic data.
| |
Patients |
| LVEDD |
48.57 |
| LVESD |
36.49 |
| LVEDV |
111.31 |
| LVESV |
58.22 |
| IVS |
12.2 |
| LVPWD |
12.22 |
| EF% |
47.5 |
| LA |
35.5 |
| Count |
56 |
Statistical Analyses
The research data was processed with the computer programs EXCEL (Microsoft Corporation, Redmond, WA) and STATGRAPHICS plus (Manugistics, Rockville, MD) for WINDOWS. The level of significance was determined as (p<0.05). In cases with different from normal distribution, median was used (M), together with first and third quartile Q1 and Q3; (twenty-fifth and seventy-fifth percentile P25 and 75P). Multivariate regression analysis was performed too.
Results
Serum MMP-12 levels were statistically significantly lower in patients than in controls: 0.0033 (0.0022÷0.0071) vs. 0.0075 (0.0068÷0.016) (KW=7.37; p=0.006) (Table 3 and Figure 1). MMP-12 showed correlation with treatment with ACE (r=0.48; p=0.005) and ARB (r=-0.33; p=0.05); BMI (r=0.33; p=0.05), MMP-12 and grade of AH (r=0.28; p=0.03); MMP-12 and statin (r=-0.30; p=0.02). TIMP-3 levels in patients were lower- 5.051 (2.062÷10.463) than controls 6.460 (1.007÷12.520) (p<0.05), but not significantly (Table 4 and Figure 2). TIMP3 showed correlation with grade of AH (r=0.85; p=0.02) and PLVW (r=-0.40; p=0.03).
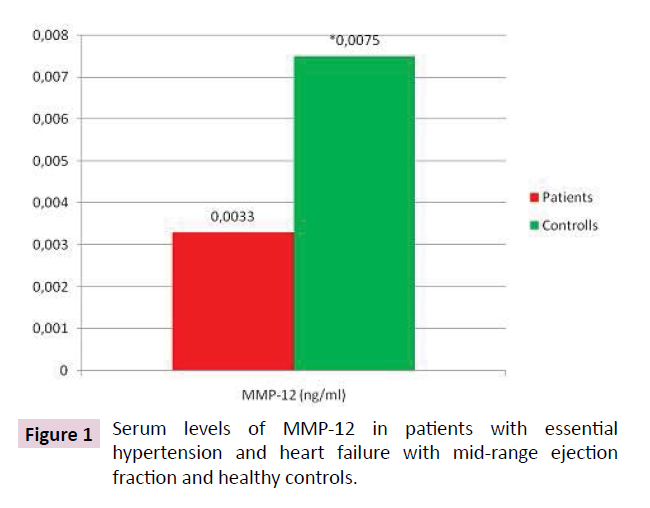
Figure 1: Serum levels of MMP-12 in patients with essential hypertension and heart failure with mid-range ejection fraction and healthy controls.
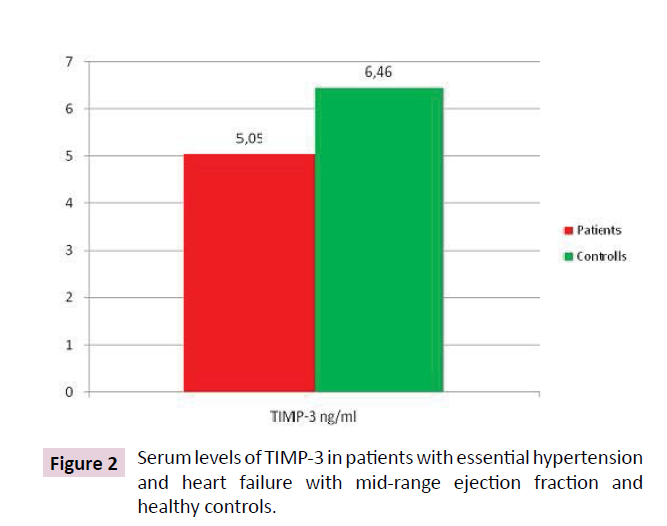
Figure 2: Serum levels of TIMP-3 in patients with essential hypertension and heart failure with mid-range ejection fraction and healthy controls.
Table 3 Serum levels of MMP-12 in patients with essential hypertension and heart failure with mid-range ejection fraction and healthy controls.
| |
MMP-12 (ng/ml) |
Comparison with |
| M÷(Q1??Q3) |
| Patients |
0.0033 |
Controls |
| (0.0022 ÷ 0.0071) |
p=0.006 |
| Controls |
0.0075 |
Patients |
| (0.0068 ÷ 0.016) |
p=0.006 |
Table 4 Serum levels of TIMP-3 in patients with essential hypertension and heart failure with mid-range ejection fraction and healthy controls.
| |
TIMP-3 (ng/ml) |
Comparison with |
| M÷(Q1??Q3) |
| Patients |
5.051 |
Controls |
| (2.062 ÷ 10.463) |
NS |
| Controls |
6,460 |
Patients |
| (1.007 ÷ 12.520) |
NS |
Discussion
Data in literature about MMP-12 and TIMP-3 turnover in patients with essential hypertension and heart failure with mid-range ejection fraction is still insufficient. The MMP-12/TIMP-3 profile in our study (↓MMP-12 and ↓TIMP-3) is corresponding with results of Hee et al. [26-28].
Hee et al. [26] test the hypothesis that the activity of enzymes degrading the extracellular matrix in hypertensive patients are abnormal, and that the treatment of hypertension will normalize these abnormalities. Authors measured serum levels of metalloproteinase MMP-9, and its inhibitor, tissue metalloproteinase inhibitor (TIMP-1). Thirty-two patients with untreated hypertension (BP 168/96) had significantly lower levels of both MMP-9 and TIMP-1 when compared to 24 matched normotensive controls (BP 123/80) (P<0.001). There was no significant correlation between MMP-9 and TIMP-1 levels (P>0.2). Levels of MMP-9 and TIMP-1 were not significantly altered after 2 months of antihypertensive treatment of 29 patients despite mean blood pressure falling from 170/96 to 143/85 mmHg (P<0.001). Authors suggest that the proteolytic activities of MMP- 9 and TIMP-l are depressed in hypertensive patients and were not significantly affected by short-term antihypertensive treatment.
“The hypothesis that plasma concentrations of matrix metalloproteinase- 2 (MMP-2) and matrix metalloproteinase-9 (MMP-9), two enzymes that share similar substrate specificity (collagen type IV and V), possibly related to vascular remodelling, are altered in essential hypertension [27,28]. The second aim of the study was to assess whether chronic antihypertensive treatment with the calcium channel blocker amlodipine would normalize these alterations. Authors findings suggest that plasma concentrations of active MMP-2 and MMP-9, mainly related to vascular extracellular matrix metabolism, are depressed in patients with essential hypertension. Moreover, after six month treatment with amlodipine MMP-9 plasma levels normalize. The hypothesis that antihypertensive treatment may modulate collagen metabolism remains to be determined by further studies.”
Goncalves et al. [29] found that the plasma level of MMP-7 and -12 are elevated in type 2 diabetes mellitus, associated with more severe atherosclerosis and an increased incidence of coronary events. These observations provide clinical support to previous experimental studies, demonstrating a role for these MMPs in plaque development, and suggest that they are potential biomarkers of atherosclerosis burden and cardiovascular disease risk.
Data of Rugmani et al. [30] showed that “early matrix metalloproteinase-12 inhibition worsens post-myocardial infarction cardiac dysfunction by delaying inflammation resolution in mice. Their results reveal a novel protective mechanism for MMP-12 in neutrophil biology. Post-myocardial infarction, MMP-12i impaired CD44–HA interactions to suppress neutrophil apoptosis and prolong inflammation, which worsened LV function.”
Data for MMP-12 expression in mice and men are controversial. Despite that, evidence from both species suggests that “MMP- 12 might be a good candidate for drug therapy to prevent MI. Furthermore, selective MMP-12 inhibitors are available and have been used safely in phase II clinical studies in pulmonary disease. Even so, deeply seated MMP-12 positive macrophages might be difficult to inhibit and therefore a surrogate marker of efficacy, perhaps based on molecular imaging, might be needed as a precursor to any outcome trial” [31].
In Ciccone et al. [32] reviewed current data for using a novel cardiac bio-marker called Suppression of tumorigenicity 2 (ST2) in detecting the early stages of cardiovascular diseases and/or their progression. “Soluble ST2 is a blood protein confirmed to act as a decoy receptor for interleukin-33. It seems to be markedly induced in mechanically overloaded cardiac myocytes. Thus, HF onset or worsening of a previous chronic HF status, myocardial infarct able to induce scars that make the myocardium unable to stretch well, etc. are all conditions that could be detected by measuring blood levels of soluble ST2”. The possible role of ST2 derived-protein as an early marker of cardiovascular diseases, above all in acute decompensated and chronic heart failure are supported by data [33] who enrolled 150 patients suffering from acute decompensated HF in order to evaluate the predictive 90- day mortality power of sST2 percent change as compared to wellestablished heart failure biomarkers such as BNP or NT-proBNP. ROC analysis demonstrated the comparable predictive value of sST2 and NT-proBNP changes in predicting 90-day mortality (sST2 AUC=0.78 CI=0.69 to 0.88 vs. NT-proBNP AUC=0.78 CI=0.67 to 0.90) rather than BNP (AUC=0.67 CI=0.56 to 0.79). The optimization of the statistical analysis led physicians to consider a reduction of more than 15.5% as the best cut-off value able to identify acute decompensated HF patients at high cardiovascular risk. Some authors demonstrated that a sST2 ratio (i.e., the ratio between sST2 two week values and sST2 baseline values)>0.75 would be able to really predict cardiac event rates independently from others confounding factors [34]. The predictive value of sST2 still persists at 1-year follow-up: the higher the sST2 plasma levels, the higher the mortality rates at 1-year, independently of possible confounders, remaining associated to that of BNP (rs=0.317; 95% CI, 0.157 to 0.460; p<0.001) [35]. The combination of sST2, NTproBNP, BNP, renal function and C-reactive protein values was able to increase the AUC for 1-year mortality prediction till 0.80 in a statistically significant manner [36]. Such results were confirmed by further studies about this matter which made reliable sST2 as a real marker of acute HF and/or a good mortality predictor in the same de-compensated patients [37].
One of the first attempts to define the role of sST2 in chronic HF was realized in the neurohormonesub-study of the Prospective Randomized Amlodipine Survival Evaluation 2 (PRAISE-2) study [38], i.e., a multicenter, randomized, double-blinded, placebocontrolled study evaluating the effects of amlodipine 10 mg/day on survival of patients suffering from congestive HF due to nonischemic conditions. sST2 was significantly higher in patients with severe HF (0.24 ng/mL, CI: 0.16–0.70) than controls (0.14 ng/ mL, CI: 0.13–0.17; p < 0.0001). Scatter plots analysis suggested that sST2 values were related to BNP (r=0.36, p <0.0001), proatrial natriuretic peptide (ProANP) (r=0.36, p<0.0001), and norepinephrine (r=0.39, p <0.0001) concentrations. Nevertheless, sST2 was not able to predict mortality by itself in such a category of patients: multivariate analysis confirmed that the sST2 ratio (defined as the change in sST2 from baseline to 2 weeks treatment) was an independent predictor of mortality and/or transplantation (p=0.048), above all if associated with BNP (p<0.0001) and Pro- ANP (p<0.0001) evaluation. Penn Heart Failure Study (PHFS), a multicenter, prospective cohort study involving chronic heart failure patients suffering from different range of disease severity showed interesting results [39]. Far from the severe HF patients of PRAISE-2, those from PHFS demonstrated that sST2 evaluation had a predictive 1-year mortality/transplantation rate similar at NT-proBNP one: the ROC curves showed comparable predictive values between the two biomarkers (sST2 AUC: 0.75, 95% CI, 0.69 to 0.79 vs. NT-proBNP AUC: 0.77, 95% CI, 0.72 to 0.81; p=0.24). Furthermore, by combining sST2 and NT-proBNP evaluations, the AUC reached a 0.80 (95% CI, 0.76 to 0.84) value. Ky et al. [40] included the sST2 evaluation in a multi-markers score to be adopted in chronic HF patients in order to depict the mortality risk of such a category of patients. The new multi-markers model for chronic HF patients was superior than the Seattle Heart Failure Model (SHFM): the multi-markers score involving sST2 evaluation in combination with high-sensitivity CRP (hsCRP), myeloperoxidase (MPO), BNP, soluble fms-like tyrosine kinase receptor-1 (sFlt-1), troponin I (TnI), creatinine, and uric acid reached an AUC higher than that of SHFM (AUC multi-markers score [0.798, 95% CI 0.763–0.833 vs. AUC SHFM: 0.756, 95% CI 0.717–0.795)] at 1 year. But the combination of SHFM to the multi-markers allowed an improvement in 1-year mortality prediction up to an overall AUC of 0.803, 95% CI 0.769–0.837, p=0.003.
In our study we found that serum MMP-12 levels were statistically significantly lower in patients, than in controls. MMP-12 showed correlation with treatment with ACE (r=0.48; p=0.005) and ARB (r=-0.33; p=0.05); BMI (r=0.33; p=0.05), MMP-12 and grade of AH (r=0.28; p=0.03); MMP-12 and statin (r=-0.30; p=0.02). TIMP-3 levels in patients were lower than controls 6.460 (1.007÷12.520) (p<0.05), but not significantly. TIMP3 showed correlation with grade of AH (r=0.85; p=0.02) and PLVW (r=-0.40; p=0.03).
However, TIMPs have a much broader spectrum of targets than originally believed, which have misled our understanding of TIMPs in cardiac remodelling. “It has become increasingly clear that MMP-dependent and -independent mechanisms of TIMPs coexist, adding to the complexity of their biological role. Mounting evidence points towards an authentic signalling capacity for TIMPs distinct from their MMP-inhibitory activity, that seem to play an important role in the regulation of apoptosis, cell survival, growth, migration, differentiation, angiogenesis, inflammation and overall ECM remodeling, and may play a central role in the process of cardiac remodelling” [41,42].
The complex role of TIMP-3 is described by “the opposing observations that were reported with TIMP-3, influencing both fibroblast proliferation and apoptosis, indicate that TIMP-3 could exert different in vivo biological functions depending on the time and localization of its expression after tissue injury” [43].
“TIMP-3 has the ability to inhibit capillary morphogenesis in vivo and endothelial cell migration in vitro, through its interaction with vascular endothelial growth factor receptor-2 (VEGFR-2) and directly antagonizing the binding of VEGF, but not fibroblast growth factor-2 (FGF-2), and thereby blocking angiogenesis” [44].
Growing evidence suggests possible TIMP-3 role in the progression of vascular diseases, independently from its ability to inhibit MMPs. “In vitro and in vivo adenoviral overexpression of TIMP- 3, but not TIMP-1 or -2, promoted apoptosis in vascular smooth muscle cells, an effect that – at least in the in vivo experiments – was not achieved by the use of a synthetic MMP inhibitor” [45,46]. These studies indisputably indicate that “TIMPs function as MMP independent endogenous inhibitors of angiogenesis”. Angiogenesis is a process limited to adult tissues responding to a pathologic stimulus [47,48], yet whether this “TIMP-mediated modulation of angiogenesis is counteracting the angiogenic response during adaptive remodelling or actually contributes to maladaptive cardiac remodeling, still remains to be elucidated”.
Conclusion
1. We suggest that a double inhibition of MMP-12 and TIMP-3 exist in patients with essential hypertension and heart failure with mid-range ejection fraction. This means more serious affecting of ECM metabolism, possibly related to vascular remodeling. In result we observe decreased proteolytic function of MMP-12 in hypertonic patients with long lasting ant-hypertensive treatment. Accumulation of incorrectly cross-linked, immature and unstable elastin-degradation products may occur in result of decreased TIMP-3 levels. This process could lead to deposition of these products in vascular wall. Decreased MMP-12 levels might lead to slowdown in elastin degradation and delayed clearance of these immature and unstable elastin-degradation products.
2. TIMP-3 and MMP-12 might be eventually used as markers for control of AH. In support of that state, our results showed correlation between serum TIMP-3, MMP-12 levels and grade of arterial hypertension in patients with HfmrEF.
3. Serum MMP-12 and TIMP-3 levels might be used more likely as biomarkers for monitoring development and progression of hypertensive mediated organ damage, than prognostic markers for deterioration of heart failure with mid-range ejection fraction. We suggest this hypothesis, because our results show pathological elastin turnover with predominantly involvement of vascular ECM metabolism, than myocardial ECM, as we initially expected.
Research Limitations Section of Study
The small sample size is a limitation of the study design. Although these findings should be confirmed in a larger study, our data show correlation between serum TIMP-3, MMP-12 levels and grade of arterial hypertension in patients with HfmrEF. TIMP-3 and MMP-12 might be eventually used as markers for control of AH. However further examination and prospective studies are needed to clarify whether MMP-12 and TIMP-3 are related with development and progression of hypertensive mediated organ damage.
Conflicts of Interest
The authors do not declare any conflict of interest.
Funding Statements
This work was supported by the Centre of Scientific Research of Medical University, Pleven [project number 12/2016].
References
- National Clinical Guideline Centre (2010) Chronic heart failure: National clinical guideline for diagnosis and management in primary and secondary care: Partial Update. Guidance: National Institute for Health and Clinical Excellence.
- Munzel T, Gori T, Keaney J, Maack C, Daiber A (2015) Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Euro Heart J 36: 2555-2564.
- Brown RD, Ambler SK, Mitchell MD, Long CS (2005) The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Ann Rev Pharmacol Toxicol 45: 657-2005.
- Mann DL, Spinale FG (1998) Activation of matrix metalloproteinases in the failing human heart breaking the tie that binds. Circulation 98: 1699-1702.
- Heusch G, Libby P, Gersh B, Yellon D, Bohm M, et al. (2014) Cardiovascular remodelling in coronary artery disease and heart failure. The Lancet 383: 1933-1943.
- Hilfiker-Kleiner D, Landmesser U, Drexler H (2006) Molecular mechanisms in heart failure focus on cardiac hypertrophy, inflammation, angiogenesis, and apoptosis. J Am Col Card 48: 9.
- Mizuno T, Yau TM, Weisel RD, Kiani CG, Li RK (2005) Elastin stabilizes an infarct and preserves ventricular function. Circulation 112: I81-I88.
- Esther E, Creemers JM, Cleutjens JPM, Smits JFM, Daemen MJAP (2001) Matrix Metalloproteinase Inhibition After Myocardial Infarction A New Approach to Prevent Heart Failure? Circ Res 89: 201-210.
- ESC National Cardiac Societies (2016) ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. European Heart Journal 37:2129-2200.
- Klein T, Bischoff R (2011) Physiology and pathophysiology of matrix metalloproteases. Amino Acids 41: 271-290.
- Shapiro SD, Kobayashi DK, Ley TJ (1993) Cloning and characterization of a unique elastolytic metalloproteinase produced by human alveolar macrophages. J Biol Chem 268: 23824-23829.
- Belaaouaj A, Shipley JM, Kobayashi DK, Zimonjic DB, Popescu N, et al. (1995) Human macrophage metalloelastase. Genomic organization, chromosomal location, gene linkage, and tissue-specific expression. J Biol Chem 270: 14568-14575.
- Curci JA, Liao S, Huffman MD, Shapiro SD, Thompson RW (1998) Expression and localization of macrophage elastase (matrix metalloproteinase-12) in abdominal aortic aneurysms. J Clin Invest 102: 1900-1910.
- Proietta M, Tritapepe L, Cifani N (2014) MMP-12 as a new marker of Stanford-A acute aortic dissection. J Ann Med 46: 44-48.
- Woodruff PG, Koth LL, Yang YH, Rodriguez MW, Favoreto S, et al. (2005) A distinctive alveolar macrophage activation state induced by cigarette smoking. Am J Respir Crit Care Med. 172: 1383-1392.
- Matute-Bello G, Wurfel MM, Lee JS, Park DR, Frevert CW, et al. (2007) Essential role of MMP-12 in fas-induced lung fibrosis. Am J Respir Cell Mol Biol 37: 210-221.
- Stawski L, Haines P, Fine A, Rudnicka L, Trojanowska M (2014) MMP-12 deficiency attenuates angiotensin II-induced vascular injury, M2 macrophage accumulation and skin and heart fibrosis. Plos One 9:e109763.
- Madala SK, Pesce JT, Thirumalai R (2010) Matrix metalloproteinase 12-deficiency augments extracellular matrix degrading metalloproteinases and attenuates IL-13-dependent fibrosis. J Immunol 184:3955-3963.
- Tanner RM, Lynch AI, Brophy VH, Eckfeldt JH, Davis BR, et al. (2011) Pharmacogenetic associations of MMP9 and MMP12 variants with cardiovascular disease in patients with hypertension. Plos One 6:e23609.
- N Lamblin, C Bauters, X Hermant (2002) Polymorphisms in the promoter regions of MMP-2, MMP-3, MMP-9 and MMP-12 genes as determinants of aneurysmal coronary artery disease. J Am Col Card 40: 1.
- Srikanth G, Neetu T, Utpal S, Paras KM, Natia Q, et al. (2010) MMP-2/TIMP-2/TIMP-4 versus MMP-9/TIMP-3 in transition from compensatory hypertrophy and angiogenesis to decompensatory heart failure. Archives of Physiology and Biochemistry. J Met Dis 116:2.
- Yasmin, Wallace S, McEniery CM (2004) Matrix metalloproteinase-9 (MMP-9), MMP-2, and serum elastase activity are associated with systolic hypertension and arterial stiffness. Thrombosis and Vascular Biology 25:372-378.
- Yan J, Thomson JK, Wu X, Zhao W, Pollard AE, et al. (2014) Novel methods of automated quantification of gap junction distribution and interstitial collagen quantity from animal and human atrial tissue sections. Plos One J 9:e104357.
- Yan J, Kong W, Zhang Q, Beyer EC, Walcott G, et al. (2013) c-Jun N-terminal kinase activation contributes to reduced connexin43 and development of atrial arrhythmias. Cardiovasc Res 97: 589-597.
- Westmann CA, Alves LF, Silva-Rocha R, Guazzaroni ME (2018) Mining Novel Constitutive Promoter Elements in Soil Metagenomic Libraries in Escherichia coli. Front Microbiol 9: 1-15.
- Li-Saw-Hee FL, Edmunds E, Blann AD, Beevers DG, Lip GY (2000) Matrix metalloproteinase-9 and tissue inhibitor metalloproteinase-1 levels in essential hypertension. Relationship to left ventricular mass and anti-hypertensive therapy. Int J Cardiol 75:43-47.
- Zervoudaki A, Economou E, Pitsavos C, Vasiliadou K, Aggeli C, et al. (2004) The effect of Ca2+ channel antagonists on plasma concentrations of matrix metalloproteinase-2 and -9 in essential hypertension. Am J Hypertens 17:273-276.
- Zervoudaki A, Economou E, Stefanadis C, Pitsavos C, Tsioufis K, et al. (2003) Plasma levels of active extracellular matrix metalloproteinases 2 and 9 in patients with essential hypertension before and after antihypertensive treatment. J Hum Hypertens 17(2):119-124.
- Goncalves I, Bengtsson E, Colhoun HM (2015) Elevated Plasma Levels of MMP-12 Are Associated With Atherosclerotic Burden and Symptomatic Cardiovascular Disease in Subjects With Type 2 Diabetes. Arterioscler Thromb Vasc Biol 35:1723-1731.
- Iyer RP, Patterson NL, Zouein FA (2015) Early matrix metalloproteinase-12 inhibition worsens post-myocardial infarction cardiac dysfunction by delaying inflammation resolution. Int J Card 185: 198-208.
- Newby AC (2015) Metalloproteinases promote plaque rupture and myocardial infarction: A persuasive concept waiting for clinical translation. 44-46: 157-166.
- Ciccone MM, Cortese F, Gesualdo M (2013) A Novel Cardiac Bio-Marker: ST2: A Review. Molecules 18:15314-15328.
- Boisot S, Beede J, Isakson S, Chiu A, Clopton P, et al. (2008) Serial sampling of ST2 predicts 90-day mortality following destabilized HF. J Card Fail 14: 732-738.
- Bayes-Genis A, Pascual-Figal D, Januzzi JL, Maisel A, Casas T, et al. (2010) SST2monitoring provides additional risk stratification for outpatients with decompensated HF. Rev Esp Cardiol 63:1171-1178.
- Mueller T, Dieplinger B, Gegenhuber A, Poelz W, Pacher R, et al. (2008) Increased plasma concentrations of sST2are predictive for 1-year mortality in patients with acute destabilized heart failure. Clin Chem 54: 752-756.
- Rehman SU, Mueller T, Januzzi JL (2008) Characteristics of the novel interleukin family biomarker ST2 in patients with acute HF. J Am Coll Cardiol 52: 1458-1465.
- Diez J (2008) Serum soluble ST2 as a biochemical marker of acute heart failure. J Am Coll Cardiol 52: 1466-1467.
- Weinberg EO, Shimpo M, Hurwitz S, Tominaga S, Rouleau JL, et al. (2003) Identification of serum soluble ST2 receptor as a novel heart failure biomarker. Circulation 107:721-726.
- Ky B, French B, McCloskey K, Rame JE, McIntosh E, et al. (2011) Sensitivity ST2 for prediction of adverse outcomes in chronic heart failure. Circ Heart Fail 4: 180-187.
- Ky B, French B, Levy WC, Sweitzer NK, Fang JC, et al. (2012) Multiple biomarkers for risk prediction in chronic heart failure. Circ Heart Fail 5:183-190.
- Spinale FG (2007) Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev 87:1285-1342.
- Vanhoutte D, Schellings M, Pinto Y, Heymans S (2006) Relevance of matrix metalloproteinases and their inhibitors after myocardial infarction: a temporal and spatial window. Cardiovasc Res 69:604-613.
- Vanhoutte D, Heymans S (2010) TIMPs and cardiac remodeling: ‘Embracing the MMP-independent-side of the family’. J Mol Cell Cardio 48:445-453.
- Qi JH, Ebrahem Q, Moore N, Murphy G, Claesson-Welsh L, et al. (2003) A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med 9:407-415.
- Baker AH, Zaltsman AB, George SJ, Newby AC (1998) Divergent effects of tissue inhibitor of metalloproteinase-1, -2, or -3 overexpression on rat vascular smooth muscle cell invasion, proliferation, and death in vitro. TIMP-3 promotes apoptosis. J Clin Invest 101:1478-1487.
- George SJ, Lloyd CT, Angelini GD, Newby AC, Baker AH (2000) Inhibition of late vein graft neointima formation in human and porcine models by adenovirus-mediated overexpression of tissue inhibitor of metalloproteinase-3. Circulation 101: 296–304.
- Heineke J, Auger-Messier M, Xu J, Oka T, Sargent MA, et al. (2007) Cardiomyocyte GATA4 functions as a stress-responsive regulator of angiogenesis in the murine heart. J Clin Invest 117:3198-3210.
- Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, et al. (2007) p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 446:444-448.