Keywords
Phospholipase A2; Quercetin; Fluorescence
spectroscopy; Molecular modeling
Introduction
Phospholipases A2 (PLA2s-E.C. 3.1.1.4) are a large class of
intracellular and extracellular enzyme, catalyze the hydrolysis of
sn-2 acyl bonds of sn-3-phoapholipids. Intracellular PLA2s are
often membrane associated and are involved in phospholipid
metabolism however, extracellular PLA2s are also found
abundantly in the venom of snake and insects [1]. These enzymes are the smallest (~14 kDa), stable, calcium dependent
encountered in a wide range of biological fluids, such as saliva, venom, macrophages, platelets, placenta, etc. [2,3]. Naturally
occurring PLA2-homologues in which Asp49 is changed to Lys
[4-7], Ser [8] or Ala [9] are therefore catalytically inactive. PLA2s are highly stable protein due to the presence of disulfide bonds,
which help in analysis of biochemical and structural
characterization as well as on their pathological effects and
other bioactivities. On the basis of amino acid sequence
similarities and disulphide bonding pattern, extracellular PLA2s have been classified into class I, II and III [10].
In particular, extracellular PLA2s are ubiquitous in nature,
represents the major venom component of snakes belonging to
genus Bothrops and exhibit a broad range of biological effects
including neurotoxicity, myotoxicity, cardiotoxicity, hemolysis,
anticoagulant and antiplatelet activities [11-14]. The
Phospholipase A2 (MTX-II), purified from the snake venom
Bothrops brazili investigated earlier, a Lys49- PLA2s that are
considered to be catalytically inactive due to their inability to
hydrolyze natural phospholipids [15]. For this reason it is
important to conduct detailed studies of the microenvironment
of binding sites of PLA2s in order to understand PLA2–inhibitor
interactions. Due to these characteristics, Quercetin (QCT) has
been chosen as flavonoid model.
In this work, the interaction of QCT with MTX-II has been
investigated in aqueous solutions at three temperatures under
physiological conditions. Although there have been a large
amount of studies on the interaction of QCT with BSA [16],
Ovalbumin [17], Lysozyme [18] however, the binding of QCT with
PLA2s has seldom been reported. Therefore, the main
contribution of the present work is to describe the mechanism
of interaction between QCT and PLA2s (MTX-II) at the molecular
level.
QCT is one of the widely distributed flavonoids and
abundantly found in the flowers and plants as secondary metabolites. They are a large class of naturally occurring
bioactive polyphenolic compounds, which have three rings; the
basic nucleus of a phenyl benzo (γ)pyrone, that is, two aromatic
rings (A and B) joined by a three–carbon linked (γ)-pyrone ring (C), hydroxyl, glycosyl and methoxyl are side groups (Figure 1A)
[19]. The flavonoids are poorly water-soluble pigments and are
often referred to as “nature’s biological response modifiers”,
because of their ability to modify the body’s reaction to
allergens, viruses, and carcinogens.
Figure 1: (A) Chemical structure of QCT. (B) Tridimensional
structure of Phospholipase A2 (MTX-II) [PDB: 4DCF].
They have attracted considerable attention, specifically
because of biological and physiological importance such as
modifying eicosanoid biosynthesis, anti-estrogenic,
antiprostanoid, anti-inflammatory, antithrombic effect,
antihypertensive effect, antiarrhythmic effect, antiviral and
carcinostatic properties [20]. A large number of biochemical and
molecular biological investigations have shown that
therapeutically active flavonoid, both natural and synthetic
origin, frequently binds to proteins [21,22]. Therefore, to have a
detailed description of the interaction between flavonoid and
snake venom toxin at the molecular level is the early stage on
the long pipeline of inhibitor design (Figure 1).
Biophysical and computational assays were performed in
order to elucidate the binding and the interactions responsible
for stabilizing the QCT-MTX-II complex. MTX-II is catalytically
inactive due to the presence of Lys-49 in place of Asp-49, in the
catalytic site region, but have physiological role such as induce
myonectptic effect [23-25], inhibit the vascular endothelial
growth factor [26], indicating their biomedical relevance. This
would be main emphasis of the current work to get an inhibitor,
which affects these physiological roles. Though, in the solution
phospholipase (MTX-II) occur in tetramer conformation, but
essentially is formed by two dimers as reveled by DLS
experiment [15].
The monomers of the tetramer of MTX-II named as A, B, C and
D is shown at Figure 1B. In the solution the two dimers formed
from monomers of A and B are in the asymmetric unit display
different conformation states due to glycosylation by
tetraethylene glycol (TTEG), have active site like region.
However, the second dimer formed by molecules C and D have
an empty active site region. In the all monomers the microenvironments formed by amino acids have eight tyrosines,
which distributed on hydrophobic region as well as on the
hydrophilic surface and just one tryptophan in between the
dimmers interface. Moreover based upon experimental results
we also propose, through molecular docking, a structural model for understanding the molecular interactions between MTX-II
and QCT.
Materials and Methods
Preparation of stock solutions
PLA2 (MTX-II) was purified from the Brazilian snake; B. brazili
venom by Sephacryl S-100 column chromatography [15]. MTX-II
was dissolved in Tris-HCl buffer solution of 5 × 10-2 molL-1 at pH
8.0, containing 0.10 molL-1 of sodium chloride. The stock
solution of QCT was prepared in ethanol. The QCT stock solution
concentration was determined by using an extinction coefficient
of 19.95 mM-1cm-1 at 257 nm and 21.88 mM-1 cm-1 at 376 nm
using spectroscopy [27]. The stock solution concentration of
QCT was 3.51 × 10-3 molL-1.
However, MTX-II stock solution concentration was determined
spectroscopically using the extinction coefficient 18.295 M-1cm-1 at 280 nm, calculated by ExPASy-ProtParam tool [28] which was
3.21 × 10-4 molL-1 by monomer. The structure of QCT and MTX-II
has shown in Figure 1A and 1B. A titration experiment was
performed by adding small aliquots from QCT stock solution to
PLA2 solution at a concentration of 4.0 × 10-6 molL-1. The final
QCT concentration achieved during the titration was 11 × 10-6 molL-1(final ethanol concentration in buffer was <1%).
UV–Vis absorption spectrum
The UV–Vis absorption spectra were obtained by scanning the
solution on a Cary-3E spectrophotometer (Varian, Palo Alto, CA)
equipped with 1cm quartz cells at room temperature. The UVVis
absorbance spectra were recorded in the 200-500 nm range,
with an integration time of 0.333 seconds and spectral
bandwidth of 2.0 nm. It was performed the baseline subtraction
of the buffer for the final spectrum of each solution analyzed.
Fluorescence spectroscopy and titrations
The fluorescence measurements were performed at 288, 298
and 308K using an ISS PC1 steady-state spectrofluorimeter
(Champaign, IL, USA) well-appointed with a Neslab RTE-221
thermostat bath. Both excitation and emission bandwidths were
set at 4.0 nm. The fluorescence emission spectra were recorded
in the range of 290-500 nm follows at excitation at 280 nm. The
selected wavelength was chosen because it provides aggregate
excitation of tryptophan and tyrosine residues. The emission
spectrum was corrected for the background fluorescence of the
buffer. The absorbing effect produced by presence of QCT in
titration solution was minimized due to its low utilized
concentration (up to 11 × 10-6 molL-1).
Ab Initio calculation
The Gaussian 09 quantum chemical program series [29]
provided by Núcleo de Computação Científica da Universidade
Estadual Paulista (NCC/GridUNESP) was used in the calculation
of the chemical structure of QCT. The optimized geometry was
calculated for the gas phase isolated QCT molecule by using DFT/
B3LYP functional with 6-311+G (d,p) basis set. The vibrational frequency calculation was performed to check if the structures is
stable (true minimum on the potential energy surface) whereas
no imaginary frequencies should be found. The gap energy
between the highest occupied molecular orbital (HOMO) and
lowest unoccupied molecular orbital (LUMO) for QCT molecule
was calculated, as well as its molecular electrostatic potential
(MEP). The NewZMat utility of Gaussian program was used to
obtain the PDB file of the QCT molecule.
Molecular modeling calculation
The crystal structure of MTX-II (PDB ID: 4DCF) was obtained
from the Protein Data Bank [15]. The 3D structure of the QCT
molecule was obtained from Gaussian 09 program series. The
AutoDock Tools software [30] of MGL Tools 1.5.4 program was
used to prepare MTX-II and QCT by adding polar hydrogen
atoms, partial charges and atom types. The ligand rigid root was
generated automatically and all possible rotatable bonds and
torsions were defined as active. Grid maps were generated with
0.625 Å spacing and dimensions of 82 × 122 × 84 points by the
AutoGrid 4.2 program [31] for the whole dimer. The AutoDock
4.2 program was used to study the binding site between QCT
and MTX-II by applying the Lamarckian genetic algorithm (LGA)
for minimization using default parameters. Random starting
positions on the entire protein surface and random orientations
were used for the ligand. For each docking simulation, 100
different conformers were generated. The software Pymol [32]
was used to visualize the docked conformations and to locate
the interactions between QCT and MTX-II.
Results and Discussion
Fluorescence quenching rate constant of PLA2
Fluorescence quenching is related to any process that reduces
the fluorescence intensity of a sample. Quenching can be caused
by a variety of molecular interactions such as excited-state
reactions, molecular rearrangements, energy transfer, groundstate
complex formation, and collisional quenching.
Fluorescence quenching can be characterized by two main
mechanisms; static and dynamic.
These mechanisms are differentiated one each other due to
the response to temperature and viscosity dependent behavior,
or preferably with measurements of fluorescence lifetime. Since
higher temperature increases diffusion coefficients the
bimolecular quenching rate constants follow such increment,
because of the enlargement of the collisional quenching extent.
In contrast, weakly bounded complexes are dissociated at higher
temperature decreasing the quenching rate constant; hence
leading to less static quenching [30,33-35].
Fluorescence quenching data was analyzed by the Stern–
Volmer equation:
F0/F=1+ Kqτ0[Q]=1+ Ksv[Q] (1)
Where F0 and F are the steady-state fluorescence intensities
in the absence and presence of quencher, respectively. In Eq.
(1), Kq is the bimolecular quenching constant, τ0 is the lifetime of
the fluorophore in the absence of quencher and its value for MTX-II is 6.05 × 10-9s [36]. [Q] is the concentration of quencher
and KSVis the Stern–Volmer quenching constant.
In this experiment, the solution concentration of MTX-II was
maintained constant at 4.0 × 10-6 molL-1 and the QCT
concentration varied from 0 to 11 × 10-6 molL-1 with increments
of 1.0 × 10-6 molL-1. The spectra profiles, seen at Figure 2,
indicate the contribution of two intrinsic fluorophores species,
tyrosine (Tyr) and tryptophan (Trp), due to the presence of two
peaks at 305 and 350 nm, respectively [37]. The effect upon
fluorescence intensity of MTX-II with the increment of QCT
concentration is registered at Figure 2.
Figure 2: Fluorescence spectra of the MTX-II obtained with
the increment of the QCT concentration (T=298 K, pH 8.0, λex=280 nm). [MTX-II]=4.0 × 10-6 molL-1; [QCT] varied from 1
to 12 from 0 to 10 × 10-6 molL-1 with increment of 1× 10-6 molL-1. The insert corresponds to the emission spectra of
MTX-II (at the absence QCT) dependent on temperature.
As can be seen the fluorescence intensity of MTX-II decreased
in the presence of QCT concentrations, indicating that the
microenvironments of fluorophores were affected by flavonoid.
The spectra profiles also revealed that both peaks (305 and 350
nm) are simultaneously affected and regarding the individual
contribution which is different may be due to the
microenvironments where they are positioned in the protein. In
order to elucidate the response behavior of each contribution at
the absence of QCT, a set of experiments at different
temperatures were performed. The insert at Figure 2 shows two
similar spectra profiles at 288 and 298 K, while at 308 K the peak
intensity changed, while at 305 nm the intensity increased at
350 nm it decreased. Certainly such response may be related to
the slight conformational changes induced by temperature
varying differently the microenvironments where the intrinsic
fluorophores are found at MTX-II.
These uneven behaviors may also indicate that the quencher
accessibility is different depending on the regions of interaction with MTX-II [33]. As an example, the emission of tryptophan
(W68) at 350 nm illustrates that this residue under certain
condition is easier exposed to the buffer [33], and which
localization is at the dimmer interface [15]. On the other hand,
tyrosine with a total of eight residues, taking in account their
tridimensional arrangement and the spatial proximity (6-8 Å) are
dispersed in three main clusters, the first (Y21, Y24, Y103 and
Y107), the second (Y45 and Y51) and the third (Y64 and Y66),
shown in Figure S1. In face of major number and spatial
distribution, the final emission as a summation do not allow us
to infer much about the increment of the signal but confirms
that different microenvironments somehow made the quantum
efficiency of tyrosines more effective. The results of
fluorescence quenching at 288 K, within the concentration range
of QCT, are in accordance with the Stern–Volmer equation,
which calculus at both wavelength 305 nm and 350 nm obtained
the quenching constant (KSV) and which values are presented at
(Table 1). It is noticeable that the quenching constant for Trp is a
little bit higher than to Tyr, which reinforces our assumption
about the solvent accessibility. The same procedure was
repeated at 298 K and the quenching constant decreased at
both wavelength, and once again showed the same behavior.
The Stern-Volmer quenching constant obtained by the slope of
linear adjustment and is inversely correlated with temperature is
presented at Figure 3. The decrement of the KSVvalues as
temperature increases up to 298 K infers that the quenching
mechanism of the interaction between MTX-II and QCT is static.
In addition, the calculated values in Table 1 show that the binding reaction was greater than 2.0 × 1010 M-1s-1, the
maximum diffusion collision quenching rate constant of various
quenchers with biopolymers [38]. Therefore, the fluorescence
quenching mechanism of MTX-II-QCT is classified as static
quenching process.
| pH |
T(K) |
|
KSV |
Kq |
Ka |
Kb |
n |
| 8 |
288 |
Tyr |
4.4 × 104 M-1
(R=0.983) |
4.4 × 104 M-1
(R=0.983) |
|
5.8 × 104 M-1
(R=0.988) |
1.3
(R=0.988) |
| Trp |
5.8 × 104 M-1
(R=0.989) |
5.8 × 104 M-1
(R=0.989) |
|
7.4 × 104 M-1
(R=0.983) |
1.2
(R=0.983) |
| 298 |
Tyr |
6.2 × 103 M-1
(R=0.970) |
6.2 × 103 M-1
(R=0.970) |
|
0.9 × 102 M-1
(R=0.945) |
0.3
(R=0.945) |
| Trp |
1.2 × 104 M-1
(R=0.963) |
1.2 × 104 M-1
(R=0.963) |
|
1.0 × 103 M-1
(R=0.910) |
0.4
(R=0.910) |
| 308 |
Tyr |
|
|
2.3 × 105 M-1
(R=0.973) |
|
|
| Trp |
|
|
2.7 × 105 M-1
(R=0.935) |
|
|
Table 1: Stern-Volmer quenching (KSV), bimolecular quenching constant (Kq), modified Stern-Volmer quenching constant (Ka),
binding constant (Kb), and binding site interactions (n) of MTX-II-QCT complex at 288, 298 and 308 K.
Figure 3: Stern-Volmer plots for the fluorescence quenching
of the MTX-II by QCT at 288 and 298 K and at pH 8.0, for
tyrosine (Tyr; , ) and tryptophan (Trp; , ○).
At temperature 308 K, Stern-Volmer plot presents a slight
negative deviation from linearity (Figure 4). The downward
curvature on quenching of MTX-II reflects different
accessibilities to the intrinsic fluorophores in protein [33] along
the quenching process. This reinforces the conformational
changes assumption of the dimer, because the position of the
tryptophan residues (W68) at the dimer interface becomes more
exposed to the solvent, enhancing their accessibility areas to the
quencher when compared to the tyrosines. Thus, the quenching
data was analyzed by the modified Stern-Volmer equation at 308
K [39] see the insert at Figure 4.
Figure 4: Stern-Volmer plot for the fluorescence quenching of
the MTX-II by QCT at 308 K and pH 8.0, for tyrosine (Tyr; ) and
tryptophan (Trp; ). The inset corresponds to the modified
Stern-Volmer plot.
F0/ΔF=(faKa)-1[Q]-1+fa-1 (2)
Where ΔF is the difference between the fluorescence
intensity in the absence and presence of the quencher at various
concentrations [Q], fa is the fraction of accessible fluorescence
and Ka is the effective quenching constant for the accessible
fluorophores.
The response between F0/ΔF and [Q]-1 is linear, with a slope
equal to the value of (faka)-1. The value fa-1 is fixed on the
ordinate. The constant Ka is the quotient between the ordinate
and the slope. The fluorescence quenching results analyzed
when using Eq. (2) at 308 K is shown in Figure 4. The modified
Stern-Volmer constant value was 2.3 × 105 M-1 and 2.7 × 105 M-1 at 308 K for Tyr and Trp at 305 and 350 nm, respectively, as
shown in Table 1.
Analysis of binding equilibria
In the case of the static quenching mechanism, the
assumption adopted is that the binding sites are the same and
independent at the protein. For the description of the sitebinding
model:
P+nD→DnP (3)
Where P is the protein (MTX-II), D is the drug (QCT), and DnP
is the new complex molecular which binding constant is Kb. Here
Kb is:
Kbn=[Dnp]/[Pu][Du]n (4)
Considering that all amount of protein and drug is P0 and D0,
respectively, then [P0]=[Pu]+[DnP] and [D0]=[Du]+[DnP], so that
the relation between the fluorescence intensity and the
unbound protein (Pu) is given by [Pu]/[P0]=F/F0 and therefore
[40]:
Log(F0-F)/F=nlogKb+nlog(1/([D0]-(F0-F)[P0]/F)) (5)
Where Kb is the binding constant for a site and n is the
number of binding sites per protein (MTX-II). Figure 5 shows the
binding equilibrium plots for the fluorescence quenching of
MTX-II by the QCT molecule at 288 and 298 K. The dependence
of Log(F0-F)/F on the value of log(1/([D0]-(F0-F)[P0]/F)) is linear,
with a slope equal to the value of n and the value nlogKb fixed on the ordinate. For the system MTX-II - QCT, Kb and n value at
288 and 298 K are shown in Table 1. It may be observed that the
number of binding sites between MTX-II and QCT at 288K is
approximately equal to 1.0 (Tyr, 1.3; Trp, 1.2), indicating that
there is probability of only one QCT molecule for each MTX-II
molecule, in both signals (tyrosine and tryptophan). The data
shown in Table 1 indicates that the complex formed at 288 K is
more stable than to the complex formed at 298 K. It is also
observed that at 288 K the binding constant is magnitude orders
higher than the complex at 298 K. This means that the more QCT
molecule is carried out if the temperature increases as similar in
case of MNP-BSA complex [41]. All the correlation is greater
than 0.9 indicating that the interaction between MTX-II and QCT
is in good agreement with site binding model. Table 1 also
shows the result of Kb decreasing in correlation with the
temperature rising, which is in agreement with trends of KSV as
mentioned previously (at 288 and 298 K). This fact indicates that
the interaction between QCT and MTX-II weakens when the
temperature rises, resulting in the reduction of stability of QCTMTX-
II complex (static process).
Figure 5: Double-log plots for the fluorescence quenching of
the MTX-II by QCT at 288 and 298 K and pH 8.0, for tyrosine
(Tyr; , ) and tryptophan (Trp; , ○).
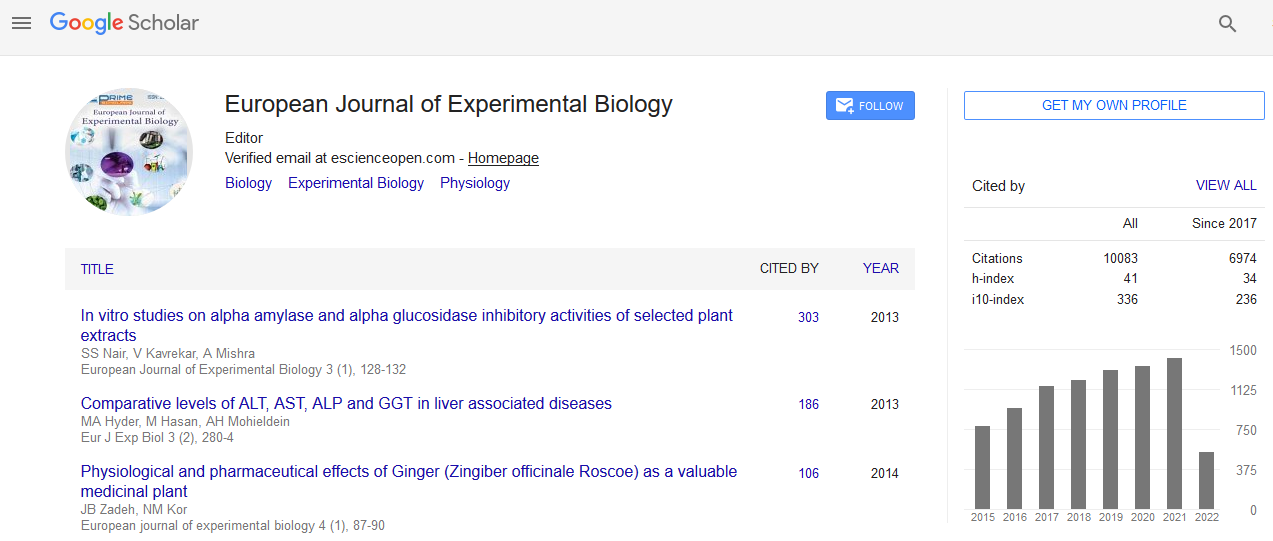
Thermodynamic analysis
The interaction forces between drugs and biomolecules may
include electrostatic interaction, multiple hydrogen bonds, van
der Waals interactions, hydrophobic and steric contacts within
the antibody-binding site, etc. [42]. The thermodynamic
parameters, such as enthalpy, entropy and free energy changes
are the main evidences to determine the binding mode.
Therefore, the thermodynamic parameters depending on
temperatures were analyzed in order to better characterize the
driving forces between MTX-II and QCT, which were calculated
from the van’t Hoff plots.
If the enthalpy changes (ΔH) does not vary significantly within
the temperature range, both the enthalpy (ΔH) and entropy (ΔS)
changes can be evaluated from the van’t Hoff equation:
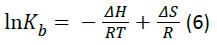
where R is the universal gas constant and Kb is binding
constant at the corresponding temperature (T). Table 2 shows
the value of ΔH and ΔS obtained from the slopes and ordinates
of the van’t Hoff relationship and lists the corresponding value
of Gibbs free energy change (ΔG) as calculated from the relation:
| pH |
|
T(K) |
ΔH(kJmol-1) |
ΔG(kJmol-1) |
ΔS(Jmol-1K-1) |
| 8 |
Tyr |
288 |
‒459.7 |
‒26.3 |
‒1,504.9 |
| 298 |
‒11.2 |
| Trp |
288 |
‒306.6 |
‒26. 9 |
‒971.3 |
| 298 |
‒17.2 |
Table 2: Thermodynamic parameters of MTX-II-QCT complex at
288 and 298 K and pH 8.0.
ΔG= ΔH-TΔS (7)
The values of entropy and enthalpy changes attained for the
binding site from the slopes and ordinates at the origin of the
fitted lines are summarized in Table 2. The negative value of
Gibbs free energy changes indicates the spontaneity of the
binding of QCT with MTX-II. Based on sign and magnitudes of
the thermodynamic parameters shown in Table 2, it is possible
to discuss the individual type of contribution of the interactions
that may occur in the complex MTX-II and QCT. It may be noted
that the interaction involving the QCT with MTX-II binding sites
is apparently sensitive to temperature. A comparison between
enthalpy and entropy change magnitudes will define whether
the hydrophobic or the electrostatic contribution will play an
important role in the stabilization of the complex. As reported,
positive values for both ΔH and ΔS imply typical hydrophobic
interaction, while negative values imply the van der Waals forces
and hydrogen bonding [16]. Based on studies of water structure,
a negative entropy change is generally considered as evidence
for hydrophilic interactions and transfer of uncharged
hydrophobic molecular form water to macromolecules has
generally been considered to be entropic [43].
The entropy and enthalpy change values obtained by the
fluorescence quenching intensity which assign the contribution
of the amino acids residues Tyr and Trp were negative and
summarized in the Table 2. The negative entropy and enthalpy
changes arise from non-bonded (Van der Walls) interactions and
hydrogen bond formation accompanying association of QCT with
MTX-II. Thus hydrogen bonds formed in a low dielectric
environment such as parts of the contact areas between MTX-II,
which are inaccessible to water or the QCT binding sites in the
interior of an enzyme could collectively make a substantial
negative contribution to entropy and enthalpy changes [44] as
similar in case of interface between the subunits of the α-
chymotrypsin dimmer [45] and interaction between S-peptide
with the S-protein of Ribonuclease [46]. The estimated result
reveals that all the interactions play almost equivalent role in
the complex QCT and MTX-II.
Distance measurement between MTX-II and QCT
Fluorescence resonance energy transfer (FRET) is a
nondestructive spectroscopic method has been used for
measuring molecular distances and relative angular orientation
of fluorophores in the biological and macromolecular systems. In
general, FRET happens when the emission spectrum of a
fluorophore (donor) overlaps with the absorption spectrum of
another molecule (acceptor). In addition, according to Förster’s
non-radiative energy transfer theory both, the donor and
acceptor fluorophores can be totally distinct or close to the
same macromolecule and the distance between the donor and
acceptor is less than approximately 8.0 nm [47]. According to
this theory, the distance r in the binding between MTX-II and
QCT can be calculated by the equation:
E=1-F/F0=R06/(R06+r6) (8)
where, E is the efficiency of transfer between the donor and
the acceptor, r is the average distance between the donor and
the acceptor, and R0 is the critical distance when the transfer
efficiency is 50%.
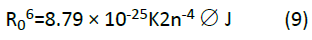
In Eq. (9), K2 is the orientation space factor, n is the refracted
index of the medium, is the fluorescence quantum yield of the
donor, J is the effect of the spectral overlap between the
emission spectrum of the donor and the absorption spectrum of
the acceptor (Figure 6), which may be calculated by the
following equation:
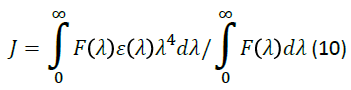
Figure 6: Spectral overlaps of the MTX-II fluorescence (A) with
QCT absorption (B) ([MTX-II]=[QCT]=4.0 × 10-6 molL-1).
Where, F(λ) is the fluorescence intensity of the fluorescent
donor in the wavelength range from λ to λ+Δλ and ɛ(λ) is the
extinction coefficient of the acceptor. In order to obtain the
distance between the donor (MTX-II) and acceptor (QCT) the
concentration of both was 4.0 × 10-6 molL-1. In this study,
K2=2/3, n=1.36, and ∅=0.093. The ∅ value was calculated by us
with the help of a lifetime of the fluorophore as reported by Alcala et al. [48]. According to Eqs. (8)-(10), it was calculated
that J=1.791 × 10-14 cm3Lmol-1, E=0.084, R0=2.57 nm and r=3.82
nm. The mean distance between donor and accepter, at which
non-radiation transfer can occur, is in the range 2.0-8.0 nm [49],
indicating that occur a possible energy transfer from MTX-II to
QCT.
Ab Initio calculations
Ab initio calculations were performed using the Gaussian
program series 09 provided by Grid UNESP. The optimized
geometry was calculated for the gas phase isolated QCT
molecule by using B3LYP functional with a 6-311+G(d,p) basis
set. Full optimization was performed with no symmetry
constraints. The calculated vibrational frequencies verified that
the structures were stable (true minimum on the potential
energy surface) whereas no imaginary frequencies were
obtained. The gap energy value between the HOMO and LUMO
molecular orbital is a molecular property and is related to an
approximation of the first electronic excitation energy [50-51].
The HOMO–LUMO energy gap of the QCT is about 3.627 eV
(ΔEgap=ΔELUMO-ΔEHOMO), which corresponds to a wavelength of
342 nm. Such wavelength characterizes the HOMO→LUMO
transition which is in the range of the absorption spectrum of
QCT in aqueous solution (Figure 6). Therefore, the results of ab initio calculation for QCT (in the gas phase) are consistent with
the absorption profile band obtained experimentally. Such
approach indicates that the optimized geometry is reasonably
accepted for the next step of the description of the interaction
between QCT and MTX-II. The molecular electrostatic potential
(MEP) of QCT (Figure 7) consists basically of the potential energy
of a proton test charge at a particular location near a molecule.
Figure 7: Molecular electrostatic potential (MEP) map of QCT
molecule calculated using the B3LYP/6-311+G(d,p) level. The
electropositive and electronegative regions are represented
by blue and yellow colors, respectively.
In our case, the MEP describes the potentially high positive
charge distribution occurs around the A and the B ring of QCT as
well have a partial negative charge. The ring C potentially has a high negative charge in comparison to A and B. Therefore, the
MEP describes equal charge distribution of QCT, which would
interact with MTX-II in the context of electrostatic potential to
perform the binding interactions.
Molecular modeling calculations
The crystal structure of MTX-II obtained from the Protein Data
Bank (PDB ID: 4DCF) [15]. QCT geometry optimized (in the gas
phase) obtained from ab initio calculation were employed to
analyze the binding sites of QCT with MTX-II (Figure 8). It shows
the result of the two best binding energies out of 100 different
conformers generated by the molecular docking. The first and
largest cluster is located in between the dimer interface. For the
lowest energy pose in this cluster, MTX-II and QCT interacts
resulting in energy of -27.8 kcaLmol-1 and a related binding
constant of 7.4 × 104 M-1, by using the equation:
Figure 8: The interaction model between Quercetin (QCT) and
MTX-II, only residue are around 3.1 Å of the ligand. The
residues of the MTX-II are presented using stick (blue for
chain A and magenta for chain B). (A) The QCT docking at the
interface of dimer. (B) The QCT docking at the active site like
region of chain B. The hydrogen bond and salt bride are
represented using yellow broken line.
K=exp(-Gb/RT) (11)
Where, K is the theoretical binding constant, R is the universal
gas constant, T is the temperature and Gb is the binding energy.
The K value obtained at 298 K in comparison with the
experimental value at the same temperature, shown in Table 1,
is in good agreement.
The binding site is in the interface region, where W68 of both
monomers are in close proximity. However, this conformation is
stabilized by three potential hydrogen bonds and three salt
bridges between QCT and MTX-II chains. Two hydrogen bonds
formed in between the carbonyl oxygen of G14 at chain B and
carbonyl oxygen of Q11 at chain A with 4ˈ-OH and 3-OH
hydrogen of QCT, respectively. The third hydrogen bond formed
in between side chain oxygen of Q11 at chain B with 5-OH
hydrogen of QCT. The interaction is also stabilized by three salt bridges; two salt bridges formed with single esoteric oxygen of
γ-pyrone ring C of QCT with carboxyl oxygen of Q11 at chain A
and side chain oxygen of Q11 at chain B of MTX-II, respectively.
The third salt bridge formed from oxygen of 3ˈ-OH of QCT with ζ
–NH3+ (amino group) hydrogen of Lys 7.
The second largest cluster is situated within the active site like
region formed by helix H1 and H2. The lowest energy pose in
this cluster is -25.8 kJmol-1 resulting in a theoretical binding
constant of 3.4 × 104 M-1. This conformation is stabilized by
three potential hydrogen bonds and one salt bridge. The
hydrogen bonds occur in between carbonyl oxygen of P17, G29
and C44 of MTX-II with 7-OH, 3ˈ-OH and 3-OH hydrogen of QCT,
respectively. The QCT binding in the active site like region are
supported by salt bride in between 3-OH oxygen of QCT with δ1-
NH hydrogen of His 47. The salt bridge formed with H47, which
can be responsible for a physiological role of MTX-II, so this
result inform that QCT can be used as an inhibitor for MTX-II.
The two binding sites found in docking analysis are assumed to
be present in Phospholipase A2. This is in line with the results
achieved by Ohno et al. [52] about the secreted Phospholipases.
Conclusion
In this paper, the interaction of QCT with MTX-II has been
investigated in vitro as well as in combination with ab initio and
molecular modeling calculations. These assays indicate that QCT
can bind to MTX-II in between the dimer interface and to the
active site like region. The most probable quenching mechanism
for the interaction between QCT and MTX-II is static process,
due to the cutback on the Stern-Volmer quenching constant
(KSV) up to temperature 298K, but at 308K the fluorescence
quenching process reflects the difference of accessibility of the
quenchers to the intrinsic fluorophores. The thermodynamics
parameters indicates that the interaction is spontaneous (ΔG<0),
and negative entropy and enthalpy data support hydrogen bond
formation. The distance (r) between donor and acceptor is
obtained according to Forster non-radioactive energy theory
(FRET). Spectroscopic experiments and molecular modeling
assays indicated that hydrophobic surface supports binding of
QCT at two different sites. The binding study of QCT to MTX-II is
of great importance to understand chemical-biological
interactions for future drug design, pharmacology, and
biochemistry studies. Furthermore, these assays are expected to
provide more information about the interactions of the protein
MTX-II with natural product in view of an application as a
therapeutic drug.
Acknowledgements
The authors would like to acknowledge the financial
assistance from CAPES/CNPq/CNPq in the form of research
fellowship to RK/IPC/AU respectively. Financial assistance from
FAPESP and TWAS also acknowledged.
References
- Arni RK, Ward RJ (1996) Phospholipase A2-A structural review. Toxicon 34: 827-841.
- Van Deenem LLM, de Haas GH (1963) The substrate specificity of phospholipase A2. Biochem Biophys Acta. 70:538-553.
- Waite M (1990) Phospholipase, enzymes that share a substrate class. Adv Exp Med Biol 279: 1-22.
- Maraganore JM, Merutka G, Cho W, Welches W, Kezdy FJ, et al. (1984) A new class of phospholipase A2 with lysine in place of aspartate. J Biol Chem 259: 13839-13843.
- Francis B, Gutierrez JM, Lomonte B, Kaiser II (1991) Myotoxin II from Bothrops asper (Terciopelo) venom is a lysine 49 phospholipase A2. Arch Biochem Biophys 284: 352-359.
- Homsi-Brandenburgo MI, Queiroz LS, Santo-Neto H, Rodrigues-Simoni L, Giglio JR (1988) Fractionation of Bothrops juraracussu snake venom: partial chemical characterization and biological activity of bothropstoxin. Toxicon 26: 615-627.
- Ward RJ, Monesi N, Arni RK, Larson RE, Paco-Larson ML (1995) Sequence of a cDNA encoding bothropstoxin I, a myotoxin from the venom of Bothrops jararacuss. Gene 156: 305-306.
- Krizaj I, Bieber AL, Ritonja A, Gubensek F (1991) The primary structure of ammodytin L, a myotoxic phospholipase A2 homologue from Viperu ammodytes. Venom. Eur J Biochem 202: 1165-68.
- Liu CS, Chen JM, Chang CH, Chen SW, Teng CM, et al. (1991) The amino acid sequence and properties of an edema-inducing Lys-49 phospholipase A2 homolog from the venom of Trimeresus mucrosquamatus. Biochem Biophys Acta 1071:362-370.
- Renetseder R, Brunie S, Dijkstra BW, Drenth J, Sigler PB (1985) A comparison of the crystal structures of phospholipase A2 from bovine pancreas and Crotulus utrox venom. J Biol Chem 260: 11627-11636.
- Chwetzoff S, Tsunasawa S, Sakiyama F, Menez A, Nigexine (1989) A phospholipase A2 from cobra venom with cytotoxic properties not related to esterase activity. Purification, amino acid sequence, and biological properties. J Biol Chem 264: 13289-13297.
- Kini RM (2005) Structure-function relationship and mechanism of anticogulant phospholipase A2 enzymes. Toxicon 45: 1147-1161.
- Schaloske RH, Dennis EA (2006) The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta 1761: 1246-1259.
- Kang TS, Georgieva D, Genov N, Murakami MT, Sinha M, et al. (2011) Enzymatic toxins from snake venom: structural characterization and mechanism of catalysis. FEBS J 278: 4544-4576.
- Ullah A, Souza TA, Betzel C, Murakami MT, Arni RK (2012) Crystallographic portrayal of different conformational states of a Lys49 phospholipase A₂ homologue: insights into structural determinants for myotoxicity and dimeric configuration. Int J Biol Macromol 51: 209-214.
- Caruso IP, Vilegas W, Fossey MA, Cornelio ML (2012) Exploring the binding mechanism of Guaijaverin to human serum albumin: fluorescence spectroscopy and computational approach. Spectrochem Acta A 97: 449-455.
- Lu Y, Wang YL, Gao SH, Wang GK, Yan CL, et al. (2009) Interactions of quercetin with ovalbumin: Spectroscopic and molecular modeling studie. J Luminescence 129: 1048-1054.
- Wang GK, Wang LX, Tang W, Hao XX, Wang YL, et al. (2011)Binding of quercetin to lysozyme as probed by spectroscopic analysis and molecular simulation. J Fluorescence 21: 1879-1886.
- Morand C, Crespy V, Manach C, Besson C, Demigne C, et al. (1998) Plasma metabolites of quercetin and their antioxidant properties. Am J Physiol 275: 212-219.
- Formica JV, Regelson W (1995) Review of the biology of quercetin and related bioflavonoids. Food Chem Toxicol 33: 1061-1080.
- Kaldas MI, Walle UK, Van der Woude H, McMillan JM, Walle T (2005) Covalent binding of the flavonoid quercetin to human serum albumin. J Agric Food Chem 53: 4194-4197.
- Kanakis CD, Tarantilis PA, Polissiou MG, Diamantoglou S, Tajmir-Riahi HA (2006)Antioxident flavonoids bind human sérum albumin. J Mol Struct 798: 69-74.
- Lomonte B, Tarkaowski A, Hanson LA (1993) Host response to Bothrops asper snake venom. Analysis of edema formation, inflammatory cells, and cytokine release in a mouse model. Inflammation 17: 93-105.
- Lomonte B, Lundgren J, Johansson B, Bagge U (1994) The dyanamics of local tissue damage inducecd by Bothropus asper snake venom and myotoxin II on th mouse cremaster muscle: An intreavital and electron microscopic study. Toxicon 32: 41-55.
- Lomonte B, Moreno E, Tarkowski A, Hanson LA, Maccarana M (1994) Neutralizing interactionbetween heparins and myotoxin 11, a lysine 49 phosphohpase A2 from Bothrops asper snake venom. J Biol Chem 269: 29867-29873.
- Yamazaki Y, Matsunaga Y, Nakano Y, Morita T (2005) Identification of vascular endothelial growth factor receptor-binding protein in the venom of eastern cottonmouth. A new role of snake venom myotoxic Lys49-phospholipase A2. J Biol Chem 280: 29989-29992.
- https://www.sigmaaldrich.com/etc/medialib/docs/Sigma/Product_Information_Sheet/1/q0125pis.Par.0001.File.tmp/q0125pis.pdf
- Gattiker A, Gasteiger E, Hoogland C, Duvaud S, Wilkins MR, et al. (2005) Bairoch, Protein Identification and Analysis Tools on the ExPASy Server;
(In) John M. Walker (ed): The Proteomics Protocols Handbook, Humana Press.
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, et al. (1998) Gaussian 98, Revision A. 9 Gaussian, Inc., Pittsburgh, PA.
- Sanner MF (1999) Python: a programming language for software integration and development. J Mol Graph Model 17: 57-61.
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, et al. (1998) Automated docking using a Larmarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 19: 1639-1662.
- Delano WL (2004) PyMOL software, version 0.99, USA, San Carlos.
- Lokowicz JR (1999) Principles of fluorescence spectroscopy, second ed., Kluwer academic publishers/plenum press, New York.
- Hu YJ, Li W, Liu Y, Dong JX, Qu SS (2005) Fluorometric investigation of the interaction between methylene blue and human serum albumin. J Pharm Biomed Anal 39: 740-745.
- Hu YJ, Liu Y, Wang JB, Xiao XH, Qu SS (2004) Study of the interactions between monammonium glycyrrhizinate and bovine serum albumin. J Pharm Biomed Anal 36: 915-919.
- Liu EH, Qi LW, Li P (2010) Structural relationship and binding mechanisms of five flavonoids with bovine serum albumin. Molecules 15: 9092-9103.
- Abou-Zied OK, Al-Shihi OI (2008) Characterization of subdomain IIA binding site of human serum albumin in its native, unfolded, and refolded states using small molecular probes. J Am Chem Soc 130: 10793-10801.
- Wells TA, Nakazawa M, Manabe K, Song PS (1994) A conformational change associated with the phototransformation of pisum phytochrome A as probe by fluorescence quenching. Biochemistry 33: 708-712.
- Ware WR (1962) Oxygen quenching of fluorescence in solution: an experimental study of the diffusion process. J Phys Chem 66: 455-458.
- Bi S, Ding L, Tian Y, Song D, Zhou X, et al. (2004) Investigation of the interaction between flavonoids and human serum albumin. J Mol Struct 703: 37-45.
- Seelig J, Ganz P (1991) Nonclassical hydrophobic effect in membrane binding equilibria. Biochemistry 30: 9354-9359.
- Lehrer SS (1971) Solute perturbation of protein fluorescence. The quenching of the tryptophan fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry 10: 3254-3263.
- Leckband D (2000) Measuring the forces that control protein interactions. Annu Rev Biophys Biomol Struct 29: 1-26.
- Ross PD, Subramanian S (1981) Thermodynamics of protein association reactions: forces contributing to stability. Biochemistry 20: 3096-3102.
- Vandlen RL, Tulinsky A (1973) Changes in the tertiary structure of alpha-chymotrypsin with change in pH: p4 4.2-6.7. Biochemistry 12: 4193-4200.
- Wyckoff HW, Tsernoglou D, Hanson AW, Knox JR, Lee B, et al. (1970) The three-dimensional structure of ribonuclease-S. Interpretations of an electron density map at a nominal resolution of 2 A. J Biol Chem245: 305-328.
- Weiss S (1999) Fluorescence spectroscopy of single biomolecules. Science 283: 1676-83.
- Alcala JR, Gratton E, Prendergast FG (1987) Interpretation of fluorescence decays in proteins using continuous lifetime distributions. J Biophy 51: 925-936.
- Pan BF, Gao F, Ao LM (2005) Investigation of interactions between dendrimer-coated magnetite nanoparticles and bovine serum albumin. J Magn Magn Mater 293: 252-258.
- Verheij H, Volwerk J, Jansen E, Puyk W, Dijisktra B, et al. (1980) Methylene of histdine-48 in pancreatic phospholipase A2. Role of histidine an calcium ion in the catalytic mechanism. Biochemistry 19: 743-750.
- Scott D, Sigler P (1994) Structure and catalytic mechanism of secretory phospholipase A2. Adv Protein Chem 45: 53-88.
- Ohno M, Chijiwa T, Oda-Ueda N, Ogawa T, Hattori S (2003) Molecualar evolution of myotoxic phospholipase A2 from snake venom. Toxicon 42: 841-854.